Abstract
Real-time quantitative PCR (qPCR) has become the gold standard for validating DNA microarray data and is routinely used to determine gene expression differences between a wide variety of samples. The exquisite sensitivity of the technology permits the detection of a single copy of a target gene in a sample which has led to qPCR now being used in the clinical setting to diagnose infection and disease states. In an effort to standardize the design of the associated experiments, the minimum information for publication of quantitative real-time PCR experiments (MIQE) guidelines were published in 2009. In this study, we show how qPCR can lead to erroneous conclusions regarding differences in MCM7 gene expression between normal and tumor human breast cancer samples if the key steps set out in the MIQE guidelines are not followed.
Introduction
Quantitative PCR is currently the most sensitive technique for quantification of gene expression in cells, tissue, and biological fluids. However, with the reward of sensitivity comes the risk of misinterpreted data from inadequate sample processing and handling, poor primer design and validation, and inappropriate reference gene selection. The MIQE guidelines were recently published (Bustin et al. 2009) to assist the scientific community to produce consistent, high-quality data from real-time qPCR experiments. This publication was followed by several articles that elaborate on the theme of producing high-quality and reliable data from qPCR (Bustin, 2009, Becker et al. 2010, Taylor et al. 2010).
One of the major drawbacks of qPCR is that it encompasses a series of experiments that are dependent on each other to assure solid and interpretable results such that artifactual data can easily be generated if the steps are not performed correctly (Taylor et al. 2010). The first step after experimental design and sample storage is typically the extraction of total RNA, which is followed by the reverse transcription reaction to convert the mRNA to cDNA. qPCR with validated primers, reference genes, and controls completes the steps in the process. Each of these processes generally produces some data even if the experiments are performed poorly. Errors could occur in any of these steps, leading to wrong conclusions as noted in a recent publication describing how the inappropriate selection of reference genes gives wide variation in qPCR data and misleading conclusions (Fu et al. 2009).
In this study, the minichromosome maintenance (MCM) protein, MCM7, was selected as a model target gene to investigate the importance of appropriate reference gene selection and RNA sample quality as described by the MIQE guidelines. MCM proteins have been shown to exhibit increased expression in multiple cancer types, including breast cancer (Gonzales et al. 2005). Recently, it was shown that the knockdown of MCM7 resulted in reduced tumor size and better survival in a prostate cancer mouse model (Shi et al. 2010). We used defined sample sets from breast cancer and normal patients to show how the final results and conclusions generated from qPCR data can be dramatically altered through poor application of each major step in the MIQE methodology.
Materials and Methods
RNA isolation and characterization
Ten consecutive breast cancer patients gave informed consent to provide tumor tissue samples for an institutional tumor bank at the Jewish General Hospital (Montreal). The collection of samples as well as the consent forms for this tumor bank are approved by the Institutional Review Board at the hospital. Normal and tumor breast tissue samples were ground in liquid nitrogen using a mortar and pestle. The lysates were homogenized using QIAshredder (Qiagen) and purified using RNeasy Plus Mini kit (Qiagen). The ratio of absorbance at 260 nm to 280 nm (A260/A280 ratio) was used to assess the purity of RNA samples using the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies). RNA integrity and concentration were assessed with the Experion™ automated electrophoresis station (Bio-Rad Laboratories Inc.) using the Experion RNA StdSens reagents and Experion RNA StdSens chips, according to kit instructions. The electropherograms and gels were evaluated by Experion software version 3.0.
cDNA synthesis
All RNA samples were diluted in water BioChemika (Sigma Life Science) at the same concentration and a volume corresponding to 100 ng was added to iScript™ cDNA synthesis kit reagents (Bio-Rad) for cDNA synthesis, per kit instructions. The reaction mix was incubated for 5 min at 25°C, followed by 30 min at 42°C and 5 min at 85°C and hold at 4°C. The cDNA samples were then stored at –80°C.
qPCR
Four commonly used reference genes (human GAPDH, HPRT1, 18S rRNA, and TBP) were chosen for this study. HPRT1 and TBP have been reported to have stable expression levels and GAPDH and 18S rRNA have unstable expression levels in cancer samples (Fu et al. 2009). qPCR reactions were performed on a CFX96™ real-time PCR system (Bio-Rad) operated by CFX Manager™ software (version 1.6). Cycling conditions included a polymerase activation step of 98°C for 2 min and 40 cycles of 98°C for 2 sec and annealing/extension at 60°C for 5 sec with melt curve analysis from 65 to 95°C in 0.5°C increments. The reaction mix was prepared with 5 µl of SsoFast™ EvaGreen® supermix (Bio-Rad) with 2 µl of cDNA template in a total volume of 10 µl with 300 nM final concentration of the following primers: MCM7 forward (5′-GAT GCC ACC TAT ACT TCT GCC -3′) and reverse (5′-GAT GCC ACC TAT ACT TCT GCC -3′) (Integrated DNA Technologies, San Diego, CA), TaqMan GAPDH control reagents (Applied Biosystems), and the following 20X primers: TaqMan predeveloped assay reagents for human HPRT1, 18S, and TBP (Applied Biosystems). The amplicon products for each gene were verified on a 2% agarose gel.
Results and Discussion
RNA Sample Quality and Purity
All samples gave excellent purity values with OD260/280 between 1.9 and 2.2. The RNA samples were categorized by integrity based on the Experion relative quality indicator (RQI) number, which provides a quality rating similar to the RIN number (Riedmaier et al. 2010). Figure 1 shows the virtual RNA gel image generated by the Experion and the sample concentrations, RQI, and 28S/18S rRNA ratios. Although there has been no specific threshold RQI/RIN numbers for acceptable RNA samples, studies have shown high variability in the expression data for RIN/RQI below 7 and even below 7.8 (Copois et al. 2007, Jahn et al. 2008). Four of the normal samples gave RQI values below 7.4 and the rest of the samples gave RQI values greater than 7.8.
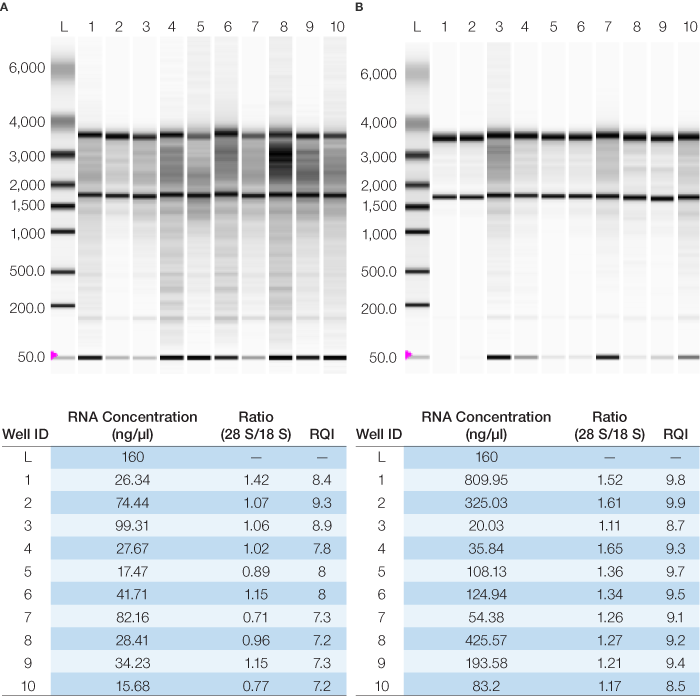
Fig. 1. Quality of RNA samples as measured by the Experion automated electrophoresis station for normal (A) and tumor (B) samples. The associated RQI, 28S/18S rRNA ratio and concentration data are shown below each gel image
Primer Validation
A pooled cDNA sample containing equal amounts of the reverse-transcribed RNA from all normal and tumor tissue samples was used to test the annealing temperature of all the primers. Primers were selected for amplicons between 100 bp and 250 bp with a target annealing temperature of 60°C. A thermal gradient qPCR reaction was programmed using the CFX96 real-time PCR system and each primer pair was tested over a 12°C temperature range to determine its optimal annealing temperature. The amplification curves were analyzed to determine the lowest Cq, which defines the optimal range of annealing temperatures for each primer pair (Figure 2A). Based on these results, a temperature of 60°C was chosen as the highest common optimized annealing temperature for all the primer pairs. Melt curve analysis for each primer pair was performed to ensure primer specificity (Figure 2B). The amplicon sizes were also verified by agarose gel electrophoresis (Figure 2C).
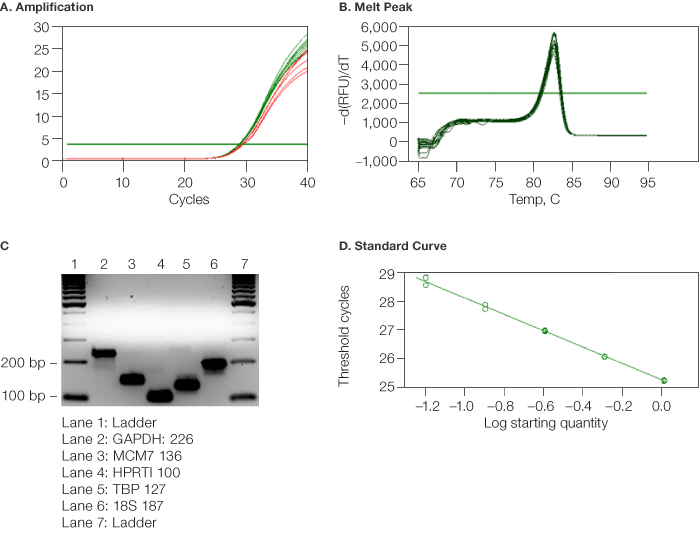
Fig. 2. Validation of MCM7 primers and samples using CFX96™ real-time PCR system (Bio-Rad). A, the amplification plots at 62°C, 61.3°C, and 50°C (![]() ) gave higher Cq values and therefore were not optimized. The rest of the curves (
) gave higher Cq values and therefore were not optimized. The rest of the curves (![]() ) converge at the lowest Cq, and the associated temperature range (50.8°C to 60°C) can be used for optimized amplification; B, the associated melt curve data from the thermal gradient were analyzed to assure good primer specificity at each annealing temperature; C, aliquots from the wells of the thermal gradient analysis were analyzed on a 2% agarose gel to assure amplicon identity (by molecular weight) and specificity: lane 1, ladder; lane 2, GAPDH (226); lane 3, MCM7 (136); lane 4, HPRTI (100); lane 5, TBP (127); lane 6, 185 (187); lane 7, ladder; D, twofold serial dilution of the pooled cDNA sample was prepared over eight dilutions. Points on the standard curve were deleted until the reaction efficiency was between 90% and 110%. Unknown samples were then diluted fourfold to the middle point of the standard curve corresponding to eightfold for MCM7.
) converge at the lowest Cq, and the associated temperature range (50.8°C to 60°C) can be used for optimized amplification; B, the associated melt curve data from the thermal gradient were analyzed to assure good primer specificity at each annealing temperature; C, aliquots from the wells of the thermal gradient analysis were analyzed on a 2% agarose gel to assure amplicon identity (by molecular weight) and specificity: lane 1, ladder; lane 2, GAPDH (226); lane 3, MCM7 (136); lane 4, HPRTI (100); lane 5, TBP (127); lane 6, 185 (187); lane 7, ladder; D, twofold serial dilution of the pooled cDNA sample was prepared over eight dilutions. Points on the standard curve were deleted until the reaction efficiency was between 90% and 110%. Unknown samples were then diluted fourfold to the middle point of the standard curve corresponding to eightfold for MCM7.
Primer and Sample Validation
The same pooled cDNA sample was then used to validate the primers for reaction efficiency at their optimal annealing temperature of 60°C (Figure 2D). Different serial dilutions were used for each gene based on the above mentioned thermal gradient–optimized Cq values as follows: Cq: 10–15: tenfold, 18S RNA; Cq: 16–20: fivefold; Cq: 21–25: fourfold, GAPDH; Cq: 26–32: twofold, HPRT1, TBP, and McM7. The cDNA samples were diluted to the mid point of the respective standard curves for each gene as follows:18S RNA: 1/10,000; GAPDH: 1/64; HPRT1: 1/8; TBP: 1/4; McM7: 1/4.
Reference Gene Validation
The reference genes (human GAPDH, HPRT1, 18S rRNA, and TBP) were tested for stability of expression across the good quality normal and tumor samples. All samples from the normal and tumor tissues were normalized for RNA concentration using the Experion automated electrophoresis station and the relative expression data (i.e., ΔCq) were then applied to the geNorm method (Vandesompele et al. 2002) to determine their stability (M value) (Figure 3) (Vandesompele et al. 2004). The least stable reference genes across the samples were 18S rRNA and GAPDH, while the most stable were HPRT1 and TBP, indicating that the use of these two genes for normalization would give the most reliable data given that their M value was well below 1.0.
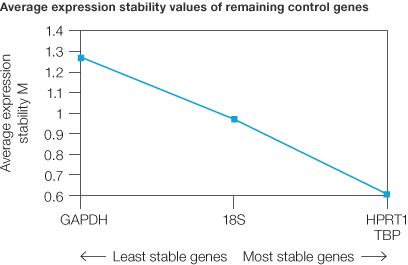
Fig. 3. Analysis of reference gene stability between normal and tumor samples. Average expression stability of control genes was calculated using the geNorm method. The two most stable reference genes are HPRT1 and TBP.
Data Analysis with Reference Gene Normalization (ΔΔCq)
Samples with RQI >7.8 were considered acceptable. All of the tumor and six of the normal samples met this criterion. Gene expression of MCM7 was then analyzed with normalization by each reference gene individually and the combination of HPRT1 and TBP, which were deemed the most stable by geNorm. The results showed that choice of reference gene for data normalization can dramatically alter gene expression data. Between normal and tumor samples, MCM7 showed either no change (when normalized against 18s rRNA), a decrease (when normalized against GAPDH) (Figure 4) or an increase in expression (when normalized using a combination of HPRT1/TBP) (Figure 4).
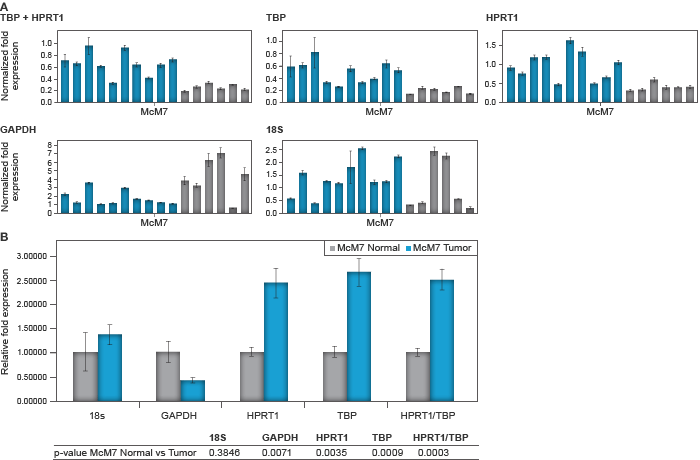
Fig. 4: Relative MCM7 expression for individual samples (A, upper panels) and associated average between the normal and tumor group normalized to each reference gene using CFX Manager software (B, lower panel). Good-quality normal samples (![]() ) versus tumor samples (
) versus tumor samples (![]() ). Error bars represent the standard error of the mean and the table beneath the chart shows the associated P values for student’s T test between normal and tumor samples.
). Error bars represent the standard error of the mean and the table beneath the chart shows the associated P values for student’s T test between normal and tumor samples.
To illustrate the effect of sample quality on the results, the four poor-quality samples from the normal tissue were analyzed against the tumor samples. Poor-quality samples were defined by an RQI less than 7.4. Relative expression of the target gene MCM7 was assessed between the poor-quality normal and tumor samples using the most stable (HPRT1/TBP) reference gene combination for normalization. No difference in gene expression was observed, as shown by P values derived using a Student’s T test (Figure 5).
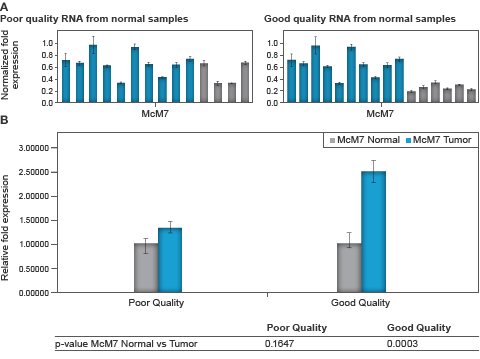
Fig. 5. Relative MCM7 expression for individual samples (A, upper panel) and associated average between the normal and tumor group contrasting poor and good quality RNA samples (B, lower panel). Normal samples (![]() ) versus tumor samples (
) versus tumor samples (![]() ). A, upper panel, bars represent the average normalized expression for three technical replicates of each sample; B, lower panel, bars represent the average relative expression for combined normal and tumor groups. Error bars represent the standard error of the mean and the table beneath the chart shows the associated P values for student’s T test between normal and tumor samples.
). A, upper panel, bars represent the average normalized expression for three technical replicates of each sample; B, lower panel, bars represent the average relative expression for combined normal and tumor groups. Error bars represent the standard error of the mean and the table beneath the chart shows the associated P values for student’s T test between normal and tumor samples.
Conclusions
For qPCR, the key steps to ensure interpretable data include: assuring good sample quality and purity; appropriate primer selection with validation by thermal gradient, melt curve, and gel analysis; primer and sample validation with standard curve and efficiency calculation; and appropriate reference gene selection with geNorm. It is imperative to diligently follow all these steps to obtain reliable data. This is demonstrated in this study with MCM7, which exhibited a significant increase in gene expression between normal and tumor samples of good RNA quality and purity when normalized using stable reference genes such as HPRT1, TBP, and their combination, in accordance with previous studies (Gonzalez et al. 2005). However, inconclusive and even opposite results were obtained with reference genes (such as GAPDH and 18S) that were least stable with this cell type and poor-quality RNA samples. This study demonstrates that inappropriate conclusions can be drawn from qPCR data if the MIQE guidelines are ignored.
References
Becker C et al (2010). mRNA and microRNA quality control for RT-qPCR analysis. Methods 50(4), 237–243.
Bustin SA (2009). Why the need for qPCR publication guidelines? — The case for MIQE. Methods 50(4), 217–226.
Bustin SA et al (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55(4), 611–622.
Copois V et al (2007). Impact of RNA degradation on gene expression profiles: Assessment of different methods to reliably determine RNA quality. J Biotechnol 127(4), 549–559.
Fu LY et al (2009). Suitable reference genes for real-time PCR in human HBV-related hepatocellular carcinoma with different clinical prognoses. BMC Cancer 9, 49.
Gonzalez MAet al (2005). Control of DNA replication and its potential clinical exploitation. Nat Rev Cancer 5(2), 135–141.
Jahn CE et al (2008). Evaluation of isolation methods and RNA integrity for bacterial RNA quantitation. J Microbiol Methods 75(2), 318–324.
Riedmaier I et al (2010). Comparison of two available platforms for determination of RNA quality. Biotechnol & Biotechnol 24(4), 2154–2159.
Shi YK, Yu YP, Tseng GC, Luo JH. (2010). Inhibition of prostate cancer growth and metastasis using small interference RNA specific for minichromosome complex maintenance component 7. Cancer Gene Ther 17(10), 694–699.
Taylor S et al (2010). A practical approach to RT-qPCR-Publishing data that conform to the MIQE guidelines. Methods 50(4), S1–S5.
Vandesompele J et al. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric aceraging of multiple internal control genes. Genome Biol 3, RESEARCH0034.
Vandesompele J et al (update: Sept. 6, 2004). GeNorm software manual. [http://medgen.ugent.be/~jvdesomp/genorm].
EvaGreen is a registered mark of Biotium, Inc.
TaqMan is a trademark of Roche Molecular Systems, Inc.
RNeasy is a registered trademark of Qiagen.
NanoDrop is a trademark of NanoDrop Technologies, LLC.


