Introduction
Laboratories involved in drug screening, diagnostics, or other medium- to high-throughput activities increasingly rely on automation to reduce time to completion, technical errors, labor, and costs. RNA extractions for use in qPCR assays are labor-intensive, time-consuming procedures that are now amenable to automation. This raises the question of whether commercially available RNA extraction kits that have been designed for low- to medium-throughput extractions using centrifugation or filtration-based methods can be used in automated high-throughput procedures.
The Aurum total RNA 96 kit (Bio-Rad Laboratories, Inc.) was adapted to a centrifugation method that allowed simultaneous extraction of two 96-well plates in the time required to complete a single plate using the standard protocol. In this article, we evaluate the use of the Aurum total RNA 96 kit in a fully automated extraction process using the KingFisher and BioSprint magnetic extraction platforms that capture targets on magnetic beads through an automated rod assembly that functions in a 96-well format (Fang et al. 2007). We adapted the Aurum total RNA 96 kit to extract RNA from human cells expressing hepatitis C virus (HCV) replicons as part of ongoing drug screening for compounds active against HCV. The same protocol was employed to help address the recent influx of clinical material associated with the H1N1 swine flu pandemic.
Methods
Cell Lines and Sample Processing
HuH-7 cells harboring an HCV replicon that expresses secreted alkaline phosphatase (SEAP) (Bourne et al. 2005) were aliquoted into 96-well plates. At selected time points after drug application, the cells were lysed using 100 µl of Aurum total RNA 96 kit-provided lysis buffer (Bio-Rad Laboratories, Inc.) containing 1% β-mercaptoethanol and stored at –80°C until use. Lysates were thawed on ice before RNA extraction. Serial ten-fold dilutions of the lysates were performed in lysis buffer.
RNA Extraction
Manual RNA extraction was performed following the Aurum total RNA 96 kit instructions with slight modifications. The DNase I digestion was increased to 20 min and samples were eluted in a volume of 150 µl. To increase throughput, the kit-provided protocol was modified by replacing the vacuum filtration steps with short spin centrifugation. This allowed two plates to be extracted at the same time. The plates were spun using a Sigma 4–15 centrifuge with a 2 x 96-well plate rotor (QIAGEN). During centrifugation, the filter plates were supported by the 96-well “grow blocks” supplied in the Aurum kit. For each wash step, the plates were spun at 4,000 rpm for 3 min. The elution step was done twice (2 x 75 µl) by spinning at 1,400 rpm for 1 min each.
Automated RNA extraction was carried out using KingFisher 96 (Thermo Fisher Scientific Inc.) or BioSprint 96 (QIAGEN) magnetic bead processing instruments fitted with deep-well magnetic head configurations in conjunction with platform-specific software. Plastic consumables, necessary for automated extraction, were purchased from VWR International. MyOne SILANE magnetic Dynabeads were obtained from Invitrogen Corporation. The computerized script to perform the Aurum kit–based 96-well extraction will be provided upon request.
Reverse Transcription (RT)–qPCR
cDNA was synthesized using an iScript™ cDNA synthesis kit (Bio-Rad). Reactions (10 µl) were assembled in 96-well PCR plates by combining 2 µl iScript reaction mix, 6.5 µl nuclease-free water, 0.5 µl reverse transcriptase, and 1 µl RNA. RT was completed using a C1000™ thermal cycler (Bio-Rad) by performing the following incubation steps: 5 min at 25°C, 30 min at 42°C, and 4 min at 85°C.
cDNA was evaluated using qPCR with primers amplifying targets within the HCV 5′ non-translated region and the human glyceraldehyde 3-phosphate dehydrogenase gene (GAPDH) (Bourne et al. 2005). TaqMan (Roche Molecular Systems, Inc.) probes were used to track the specific amplification in each duplex. Each 25 µl reaction contained 12.5 µl iQ™ supermix (Bio-Rad), 1 µl (5 µM) of each of the primers, 0.5 µl (7.5 µM) HCV-FAM probe, 0.5 µl (15 µM) GAPDH-Texas Red probe, 6.5 µl nuclease-free water, and 1 µl cDNA template. qPCR was completed in a C1000 thermal cycler equipped with a CFX96™ optical reaction module (Bio-Rad) using the following cycling parameters: 95°C for 1.5 min, followed by 40 cycles at 95°C for 20 sec and 62°C for 1 min. Data were collected at the end of each extension step. Starting quantity values were extrapolated from standard curves of plasmids harboring the PCR target generated in parallel for each run. Mathematical analyses were performed using Excel spreadsheet software (Microsoft Corporation) or Prism v4.0 analysis software (GraphPad Software Inc.).
Results
Medium-Throughput Centrifugation-Based RNA Extractions Using the Aurum Total RNA 96 Kit
The modification of the protocol from vacuum to centrifugation allowed parallel extraction of two 96-well plates in ~1.5 hr, the same amount of time required to process a single plate using the vacuum method. In order to validate the centrifugation method, sample lysates were divided equally between two 96-well plates; one was processed using vacuum and the other using centrifugation. After cDNA production, qPCR for HCV and GAPDH was performed on both sets. Analysis of the qPCR results revealed a correlation of 98% between the vacuum and centrifugation protocols, indicating that centrifugation was indistinguishable from the vacuum method for RNA production.
Development of an Automated Method for RNA Extraction Using the Aurum Total RNA 96 Kit
Before testing the Aurum total RNA 96 kit’s compatibility with magnetic bead processing, an automated procedure was developed. The assay was designed to run on either a KingFisher 96 or BioSprint 96 instrument and utilized a custom protocol generated by the associated software for each platform. The method involved sequential movement of magnetic beads via a 96-rod magnetic head between assay plates containing previously loaded Aurum kit reagents. Loading of these plates was accomplished in 15 min using a multichannel pipetor or in 6 minutes with a Tecan eight-tip liquid handler (Tecan Group Ltd.).
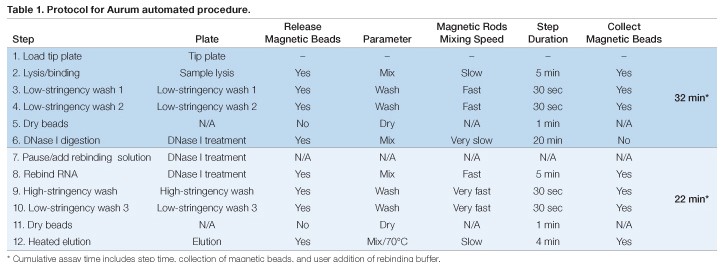
Assay plates for automated RNA extraction were prepared in advance and the KingFisher or BioSprint magnetic rods were sheathed in a disposable plastic tip comb that provided an RNase-free protective barrier during processing. These items were loaded into the instrument prior to initiating the run (Table 1). The sample plate was prepared by the addition of 100 µl of lysate (created by adding kit-provided lysis buffer to the monolayer or cell pellet) plus an equal volume of 70% RNase-free ethyl alcohol (EtOH) in addition to indicated volumes of magnetic beads. Next, 350 µl of kit-supplied low-stringency wash solution was added to each well of two deep-well plates (Table 1, Steps 3 and 4). The remaining assay plates will be described in the steps in which they were utilized below.
RNA in each well of the sample plate was first bound to the magnetic beads through automated mixing using the magnetic rods at room temperature for 5 min. The beads then were transferred to the first low-stringency wash plate. After incubation with automated mixing, the beads were transferred to the second low-stringency wash plate. This second wash ensured complete removal of protein and lysis solution chemicals before incubation with DNase I. The RNA-loaded beads were lifted out of the wash solution and air-dried for 1 min before transfer to a plate pre-loaded with DNase I solution (Table 1, Step 6). The DNase I solution was prepared by diluting 2.5 µl of reconstituted DNase I into 77.5 µl of DNase dilution buffer in each well. DNase I digestion was carried out for 20 min at room temperature. The success of DNase I digestion was evaluated by amplifying human GAPDH in the eluted RNA (no RT). The results showed only occasional DNA contamination (<100 copies/sample), similar to levels seen with the vacuum method (data not shown).
Because the conditions used for DNase I digestion could cause release of RNA from the magnetic beads, four volumes of a 1:1 ratio of Aurum total RNA lysis solution (Bio-Rad) and 70% RNase-free EtOH were manually added to the DNase I plate after digestion to favor binding of RNA to magnetic beads prior to the wash steps (Table 1, Steps 7 and 8). Subsequent high-stringency and low-stringency washes were performed as suggested in the Aurum kit protocol (Table 1, Steps 9 and 10), including an additional air-dry step (Table 1, Step 11). Elution was accomplished by incubation with agitation of the beads for 4 min in elution buffer pre-warmed to 70°C using the internal heating block in the KingFisher (or BioSprint) device (Table 1, Step 12). Using this method, RNA extraction was completed in 54 minutes.
Evaluation of Use of the Aurum Kit with Magnetic Bead Automated Processing
To assess the efficiency of automated RNA extraction, we utilized a high-throughput cell culture–based drug-screening assay using HCV replicons previously developed in our laboratory (Bourne et al. 2005). RNA was extracted from HuH-7 cells expressing HCV replicons (n = 11 replicate wells). A final well without cells served as an extraction control (lysis buffer only) for the automated procedure. qPCR for both HCV and GAPDH was then performed as described (Bourne et al. 2005). Results of this experiment indicated the presence of RNA for both qPCR targets at levels indistinguishable from the standard vacuum or centrifugation extraction methods (HCV = 5 x 105, GAPDH = 2 x 106 RNA copies). No amplification was observed in the RNA (no RT) controls indicating a lack of measurable DNA contamination. These initial results indicated that the Aurum total RNA 96 kit was compatible with automated magnetic bead extraction.
Optimization of the Automated RNA Extraction Procedure
To further validate the automation methodology and optimize the procedure, we compared automated extraction to the standard manual vacuum-based method. Sample lysates were created and then diluted to represent an input concentration of 105 cells/well in a 96-well plate prior to parallel RNA extraction by either the automated or vacuum protocol. Based on the initial success with automation, we sought to minimize costs without compromising RNA recovery by identifying the optimal amount of magnetic beads to add to each sample. When 12.5 µl or more of magnetic beads were added to the lysates, amounts of total RNA recovered were comparable to those obtained by the manual method (Figure 1), while the use of 6 µl of magnetic beads significantly decreased the RNA yield. From this experiment, 12.5 µl was identified as the optimal magnetic bead volume to use.
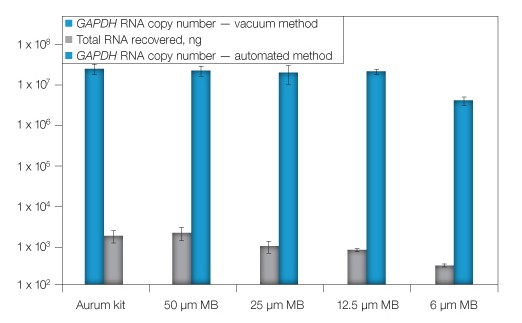
Fig. 1. Optimization of magnetic bead concentration. RNA from replicate cell lysates was extracted using the automated procedure with the addition of various quantities of magnetic beads (MB) or via the standard vacuum method (Aurum). RNA was quantified by spectrophotometry. GAPDH was evaluated by qPCR (RNA copy number). Error bars: 1 SD, n = 3, except 25 μl MB, n = 6.
Next, the dynamic range of the automated method was evaluated through a series of ten-fold serial dilutions starting with lysates of 105 HuH-7 replicon cells (Figure 2). The diluted lysates were divided evenly for extraction using manual vacuum or magnetic bead automation (using the optimal 12.5 µl of magnetic beads/extraction). The first dilution tested was expected to yield ~5 x 106 copies of HCV and ~5 x 107 copies of GAPDH, with successive dilutions providing ten-fold fewer copies of each target. As anticipated, the vacuum method provided RNA copies consistent with past studies. Comparison of the results revealed a >99% relationship (correlation coefficient r2 = 0.993) between manual and automated extractions with a lower limit of 102 (HCV) or 103 (GAPDH) across the five replicate extractions completed for each dilution. Together, these results showed the automated assay was equal to or exceeded the RNA binding capacity of the standard method over a dynamic range of approximately 107 to 102 RNA copies (Figure 2).
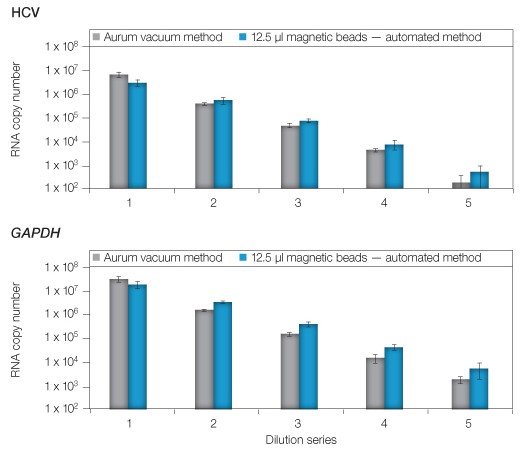
Fig. 2. Comparison of manual and automated extraction using the Aurum total RNA 96 kit. Serial dilutions of cell lysates (1–5) were matched for extraction using either the vacuum method or by automated extraction using 12.5 µl of magnetic beads. Quantity of purified RNA was evaluated via qPCR targeting HCV or GAPDH. Error bars: 1 SD, n = 3.
Application of the Automated Procedure
To demonstrate a relevant use of the automated assay, HuH-7 HCV replicon cells were evaluated by qPCR subsequent to treatment with a dilution series (0–500 µM) of a candidate antiviral designated test compound B. Triplicate wells of cells were subjected to vehicle or the selected concentrations of test compound B in 96-well plate formats. After 48 hours of treatment, RNA was extracted from cells using a standard vacuum- or magnetic bead–based protocol. HCV and GAPDH levels were evaluated by qPCR, then RNA levels, as well as the outcome of effective concentration 50% (EC50) calculations for compound B from each extraction method, were statistically compared. The EC50 values represented the concentration of drug needed to reduce the HCV RNA copy number to 50% of the untreated wells. Both methods provided statistically indistinguishable outcomes with calculated EC50 values of 0.8 µM (Figure 3, HCV). GAPDH levels also were consistent across both data sets and did not substantially decrease, indicating compound B was not cytotoxic (Figure 3, GAPDH).
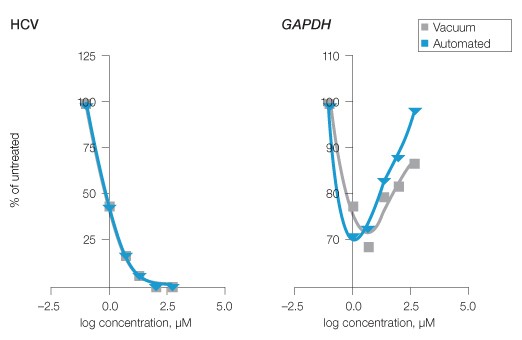
Fig. 3. Evaluation of HCV inhibition by experimental antiviral compound using manual and automated Aurum RNA extraction. HuH-7 HCV replicon cells were treated with a dilution series of an experimental antiviral compound B (0–500 µM). RNA was subsequently extracted by the standard vacuum or optimized automated protocols and qPCR for HCV and GAPDH was performed. To establish EC50 values, fourth-order polynomial curves were compared. GAPDH served as an indicator of potential test compound B cytotoxicity and allowed comparison of the procedures. Compound B HCV EC50 = 0.8 μM for both extraction methods, n = 3.
Discussion
The medium-throughput centrifugation protocol requires access to a centrifuge outfitted with a rotor capable of accommodating 96–deep-well blocks and achieving a force of ~2,600 x g. The 96-well “grow blocks” provided in the kit served to catch the flowthrough and were emptied between spins to reduce plastic requirements without causing cross-contamination. After optimization, the centrifugation protocol provided RNA from 192 samples in the time required to manually extract a single plate of 96 samples, reducing labor costs and increasing throughput. For higher throughput, we developed a protocol that took advantage of magnetic bead–based processing while successfully utilizing the reagents provided with the Aurum kit. In initial experiments, automation of the method did not result in sample cross-contamination, leading us to optimize and then validate the magnetic bead–based protocol by comparing it to standard vacuum extraction.
After optimization, we validated the automated system under a practical application of the technology that involved the screening of experimental compounds from a small molecule library that provided anti-HCV activity. In this study, the automated extraction protocol yielded activities for the selected test compound that were indistinguishable from those of the manual extraction method completed in parallel. We note that the automated extraction conditions reported here proved optimal for these experiments, but would expect that other protocols may require empirical re-optimization of magnetic bead concentration.
Conclusions
In summary, we found that increased throughput could be easily achieved with the Aurum total RNA 96 kit and determined that the optimized centrifugation and automation protocols yielded more reliable outcomes, particularly in high sample volume circumstances. Importantly, the automated Aurum kit extraction protocol took 54 minutes to complete, during which time a technician could prepare other reagents or additional experimental steps (for example, cDNA production, qPCR, etc.). Together, these results established the medium- and high-throughput capability of the Aurum total RNA 96 kit to simultaneously extract RNA from viral and eukaryotic sources with outcomes comparable to the standard vacuum method.
References
Bourne N et al. (2005). Screening for hepatitis C virus antiviral activity with a cell-based secreted alkaline phosphatase reporter replicon system. Antiviral Res 67, 76–82.
Fang X et al. (2007). Automation of nucleic acid isolation on KingFisher magnetic particle processors. JALA 12, 195–201.
BioSprint is a trademark of QIAGEN GmbH Corporation. Dynabeads and MyOne are trademarks of Life Technologies Corporation. Excel is a trademark of Microsoft Corporation. FAM is a trademark of Applera Corporation. KingFisher is a trademark of Labsystems Oy Corp. Prism is a trademark of GraphPad Software Inc. TaqMan is a trademark of Roche Molecular Systems, Inc. Tecan is a trademark of Tecan Group Ltd. Texas Red is a trademark of Invitrogen Corporation.
Notice regarding Bio-Rad thermal cyclers and real-time systems:
Purchase of this instrument conveys a limited nontransferable immunity from suit for the purchaser’s own internal research and development and for use in human in vitro diagnostics and all other applied fields under one or more of U.S. Patent Numbers 5,656,493; 5,333,675; 5,475,610 (Claims 1, 44, 158, 160–163, and 167 only); and 6,703,236 (Claims 1–7 only), or corresponding claims in their non-U.S. counterparts, owned by Applera Corporation. No right is conveyed expressly, by implication, or by estoppel under any other patent claim, such as claims to apparatus, reagents, kits, or methods such as 5′ nuclease methods. Further information on purchasing licenses may be obtained by contacting the Director of Licensing, Applied Biosystems, 850 Lincoln Centre Drive, Foster City, California 94404, USA.
Bio-Rad’s real-time thermal cyclers are licensed real-time thermal cyclers under Applera’s United States Patent Number 6,814,934 B1 for use in research, human in vitro diagnostics, and all other fields except veterinary diagnostics.
Bio-Rad’s real-time thermal cyclers are covered by one or more of the following U.S. patents or their foreign counterparts owned by Eppendorf AG: U.S. Patent Numbers 6,767,512 and 7,074,367.
Notice regarding iQ supermix:
Practice of the patented 5′ Nuclease Process requires a license from Applied Biosystems. The purchase of these products includes an immunity from suit under patents specified in the product insert to use only the amount purchased for the purchaser’s own internal research when used with the separate purchase of Licensed Probe. No other patent rights are conveyed expressly, by implication, or by estoppel. Further information on purchasing licenses may be obtained from the Director of Licensing, Applied Biosystems, 850 Lincoln Centre Drive, Foster City, California 94404, USA.


