In a healthy immune system, natural killer (NK) cells live up to their menacing name. Where other lymphocytes such as T cells or B cells target pathogens using an antibody-antigen mechanism that takes several days to activate, NK cells, the lymphocytes of the innate immune system, attack immediately, focusing their cytotoxic arsenal not on eliminating foreign invaders but on culling weakened host cells. These include tumor cells and host cells infected by viruses or microbes or affected by other forms of stress. Researchers want to better understand the mechanisms that allow NK cells perform this role, in particular how they identify stressed cells. The ability to modulate this layer of the immune system — for example, by boosting NK cells’ antitumor activity — has great medical potential.
In this video, Novák describes the promise and process of modeling natural kill receptor molecules, as well as how the NGC chromatography system helps him as he searches for the receptor structures and ligands.
NK cells’ distinctive property — what makes them “natural” killers as opposed to the antibody-antigen mechanism used by other lymphocytes—depends on their receptors. These molecules allow NK cells to recognize “self” molecules of the surface glycoproteins major histocompatibility complex (MHC) in healthy cells, as well as to identify the “missing self” molecules in the stressed or infected cells. However, many of the protein-protein interactions that make up NK cells’ precise targeting mechanisms remain a mystery.

Principal Investigator
Academy of Sciences of the Czech Republic
Petr Novák is at the forefront of the current push in research to characterize these receptors, seeking out the exact structures of these proteins. A Principal Investigator with the Academy of Sciences of the Czech Republic, Novák sees finding these structures as the first step in a process that could result in better treatments for major illnesses.
“Once we have the structure [of these receptors], then we can get the partners of these proteins, then look at the interaction of these proteins,” says Novák. “Ultimately, solving the structures of these receptors and their ligands could improve the design of biological therapies, whether [to treat] microbial infections or to fight cancer.”
More than one way to model a receptor molecule’s structure


Novák adjusts the mass spectrometry (MS) platform he uses to characterize natural killer cell receptors. X-ray crystallgraphy analyzes the protein in crystalline form; Novák was able to reveal additional details about the receptor structure by analyzing it in solution using NMR. MS-based techniques give Novák further information to incorporate into his modeling.
Getting a clear, actionable picture of receptor molecules requires the most cutting-edge and specialized structural biology techniques. While Novák’s work in this field entails a deep focus on characterizing one protein at a time, down to the subtleties of each loop and fold, he still sees this effort as an essential part of a broader effort in biology. “Even with the human genome and the other genomes sequenced, and with current proteomic data, I don’t think there’s enough information yet to understand the behavior of the overall system,” says Novák. “At this point we need to find the overview of how these proteins behave and interact with each other in the whole organism. Getting structural information is very valuable for addressing these issues.”
In 2011, Novák and colleagues reported the crystal structure of mouse NK cell receptor P1C, a common receptor found across different strains of mice and known to be involved in antitumor processes. “We are the first lab to publish the structure of NKR-P1 using X-ray crystallography,” says Novák. “And we are in the process of publishing this structure based on NMR characterization.” The way Novák uses X-ray diffraction and nuclear magnetic resonance (NMR) in tandem points to the exactness of his approach, which pays off in the molecular subtleties it reveals.
In an example of the intense detail involved in structural characterization, investigating the NKR-P1 receptor structure with NMR techniques did more than confirm the X-ray diffraction results. Novák and his colleagues were able to use NMR to discover key differences by examining the protein in solution from the structure found in the crystallized protein. These distinctions resulted from the crystal having a rigid structure while protein in solution has flexibility, especially in areas such as extended loop regions. While this might seem like a minute difference, the exact behavior of such regions — in the NKR-P1 family, one loop in particular — points to their potential ligands, and understanding these ligands in turn promises major insights into how the receptors function.
To get to this level of detail in characterizing the receptor structure, Novák and his colleagues had to devise novel combinations of techniques — in this case, using chemical cross-linking, homology modeling, and ion mobility mass spectrometry together — to elucidate a structure. So, even though the main prize is the protein structure itself, Novák is also pleased that his efforts have yielded other benefits: “It has a nice impact on the protein computing community, letting them find out that there are alternative approaches for getting three-dimensional structures, that we’re developing these techniques.”

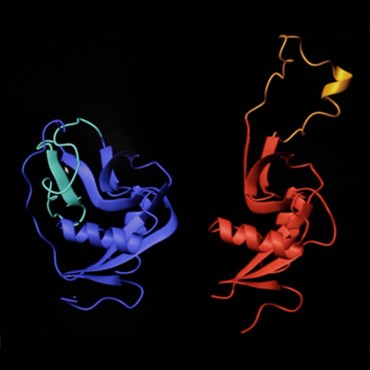
Left: Novák is a principal investigator at the Laboratory of Molecular Structure Characterization, part of the Academy of Sciences of the Czech Republic in Prague. Right: the structures of mouse NK cell receptor P1C show the subtle differences revealed by NMR (blue) and MS (red), in particular the behavior of the protein’s extended loop region in solution (turquoise loop) and in crystalline form (gold loop).
Proteins, lots and lots of correctly folded proteins
To study a protein at this level of detail, Novák needs a lot of highly pure protein. Many stumbling blocks stand in the way of obtaining usable protein. Preparing them in E. coli presented challenges because contaminants from that organism often found their way into the final product, resulting in degradation. Because Novák has to do crystallography, which requires the protein to sit for many days, even tiny amounts of proteases from the E. Coli would prevent the protein from crystallizing. Though NMR doesn’t take as many days as X-ray crystallography, it still takes enough time and occurs at high enough temperatures that contaminants from E. Coli will ruin an analysis.
Yield also poses a problem, given how much many of the chemicals involved cost. For example, to do NMR analysis Novák needs to prepare proteins doubly labeled with carbon and nitrogen isotopes. To get a worthwhile return on his lab’s investment, Novák estimated that he needed to get approximately 1 mg of pure protein for every 10 mg of inclusion bodies. “This is quite expensive,” he says. “If you don’t have a very reliable liquid chromatography system, then you can lose quite a lot of money.”
Improving the lab’s protein purification with the NGC chromatography system
Because of the many challenges involved in preparing correctly folded proteins for analysis. everything from minimizing E. coli contaminants to ensuring protein stability for mass spectrometry (MS) or NMR, Novák has to painstakingly test multiple purification strategies to arrive at a usable product.
“I used to work on so many liquid chromatography systems,” says Novák. “Through years of fighting with peptides and protein separations I learned that it’s very difficult to get pure protein. It was really painful; people in the lab spent so much time to get data.”

Novák cites the NGC chromatography system in allowing him and his students to focus more on data interpretation and less on protein preparation.

“The leak is the major enemy of a good separation,” Novák says. The instrument’s Point-to-Plumb™ lighting signals correct plumbing on the instrument front, helping Novák avoid leaks before beginning his run.
As a result of these frustrations, Novák acquired a NGC medium-pressure chromatography system from Bio-Rad. This system was designed to give researchers the flexibility to test different purification schemes and then quickly scale to higher production capacities as their needs change. Both hardware and software functionality make this possible. On the hardware side, using interchangeable modules enables multiple configurations that confer capabilities like buffer blending, multiwave detection, or other options in whatever way the researcher wishes to customize them. On the software side, integrating modules into a plug-and-play system controlled through an intuitive visual interface makes method design fast and standardized — essentially bringing chromatography into the age of touch screen devices.
This capacity to design methods quickly has cut down the time Novák needs to test different purification strategies. “I can design my method, I can see immediately that it’s correct, I can inject my sample into the system, then I can go away from the lab and I’m positive that the proteins will be there and I can pick them up for analysis when I get back,” Novák says.
Novák cites the system’s scalability for allowing him to experiment with various purification protocols. This helps him both achieve the best possible separation and save precious lab resources such as expensive isotope-labeled proteins for NMR analysis. “With our previous system, we couldn’t work in low-consumption mode,” Novák says. “Only the NGC chromatography system allows us to do an analysis with little sample consumption to find out which refolding condition is correct. Then, once we know it’s correct we can use the same system to get large-scale purification.”
Along with working in low-consumption mode, Novák also preserves his samples by troubleshooting leaks before they occur, not after. “In liquid chromatography, the leak is the major enemy of a good separation,” he says, singling out the NGC system’s Point-to-Plumb™ lighting and ChromLab™ software’s active flowpath as allies against this enemy. “If something is incorrectly plumbed, you immediately see the LED blinking, you can go there and fix it. That way you’re confident the sample will end up in the collector, not on the table.”
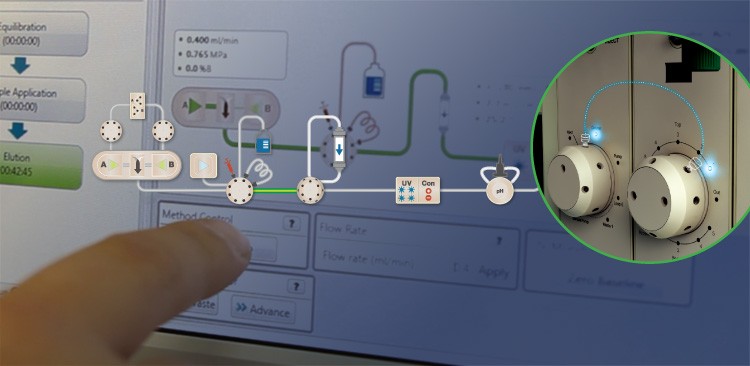
The ChromLab™ software’s active flowpath offers another tool in ensuring the system is correctly plumbed.
Currently, Novák is focused on producing NK cell receptor proteins in order to analyze their structure. However, as he looks forward Novák plans to use the NGC chromatography system in the next phase of understanding these receptors: the search for their ligands. “We can easily prepare both proteins, mix them together, run them on a gel filtration column, and see if the proteins elute as monomers or as noncovalent dimers,” he says. “Because with this system we can cut down the time for the analysis, we can try several different ligands of our receptors in a day.”
The new bottleneck in Novák’s research
“It’s a beautiful thing to have a new structure you can put into the Protein Data Bank,” says Novák. “Once it’s there everybody can use this structure to dock different ligands and see what the ligand might be. Ultimately, finding the right ligand for NK cell receptors might result in the ability to activate these cells against cancer or infections.”
Novák and his colleagues aim to characterize several more NK cell receptors in the next few years, elucidating their structures and trying different ligands for them. The NGC chromatography system has changed his lab’s priorities in achieving these goals. “Data interpretation has become the bottleneck of research,” says Novák. “Now it’s very easy for us to prepare a lot of correctly folded proteins. That means we don’t need so many students producing protein. On the other hand, we need more students interpreting the data. That’s good for us because if we can interpret more MS-based or NMR-based protein structures, we will have more answers, and we can publish more papers and show more interesting results to the public.”

