
The ability to accurately and efficiently assess cell health, whether prior to experimental treatments or as the readout of those treatments, is crucial to many experiments. In this four-part series we review common methods for evaluating viability, proliferation, apoptosis, and autophagy and briefly discuss advantages and common pitfalls.
Part I: Viability and Proliferation Assays
Viability and Proliferation
Viability and proliferation are two distinct characteristics of cells. Viability is a measure of the number of living cells in a population whereas proliferation is a measure of cell division. It should be noted that not all viable cells divide. Although proliferation can readily be interpreted as viability, absence of proliferation should not automatically be taken as a sign of cell death.
Assessing Viability and Proliferation
1. Membrane permeability
A hallmark of dead and dying cells is the breakdown of cell and nuclear membranes. Many viability assays use this characteristic of cell death to distinguish viable from nonviable cells.
Polar, cell-impermeable nucleic acid stains such as DAPI, propidium iodide (PI), and 7-AAD stain dead cells as these stains can enter only cells with compromised membranes. PI binds both RNA and DNA, while 7-AAD and DAPI interact only with DNA. These stains can be used in combination with assays that detect enzymatic activity, such as the esterase substrates discussed in the next section, or with cell permeable viability indicators to label live and dead cells simultaneously. PI, DAPI, and 7-AAD assays can be read using a fluorescence microscope, flow cytometer (Figure 1), or microplate reader.
A.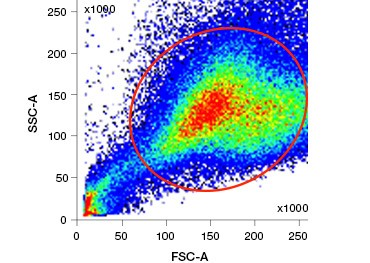
B.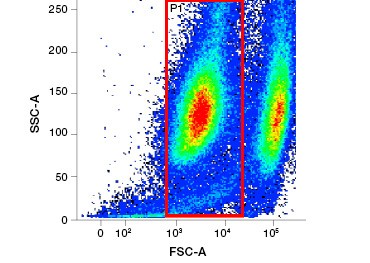
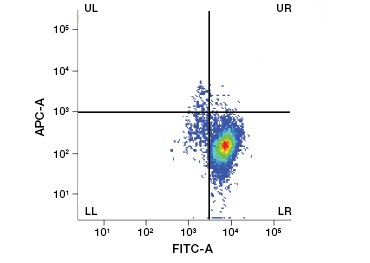
Fig. 1. Propidium iodide staining improves identification of cells of interest. Gating of live cells using forward and side scatter may not be enough to remove dead cells and non-specific binding may still be present (A). Use of viability dyes such as propidium iodide can help eliminate non specific binding (B). J774 macrophages were stained for CD11b and CD3, with the red circle and rectangle indicating the gating strategy for live cells.
Timing is critical when using these dyes because the fraction of stained cells increases during staining as cells continue to die. It should be noted that none of these dyes are fixable. For applications that require fixation, ethidium monoazides or amine-reactive protein stains are an excellent choice.
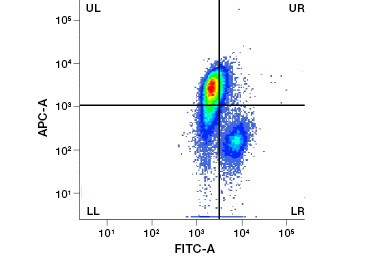
Cell impermeable protein stains are a great alternative to nucleic acid stains, as they are less toxic. Amine-reactive dyes come in a wide range of excitation and emission spectra and thus allow greater flexibility in the design of flow cytometry and microscopy panels than DNA dyes. Another advantage of these protein stains is that they are fixable (Figure 2).
A.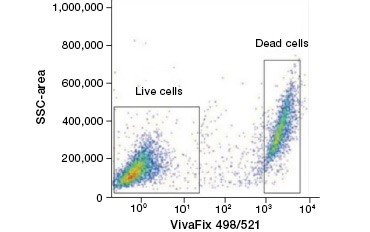
B.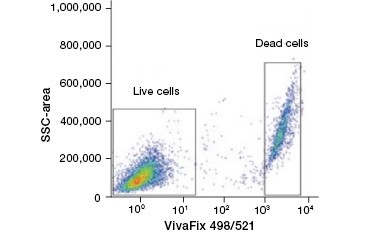
Fig. 2. Protein stains distinguish live from dead cells. Using VivaFix™ Cell Viability Assays, live and dead cell populations can be identified and subsequently analyzed in fixed (A) and unfixed (B) cells using flow cytometry.
A special consideration when using the common protein stain trypan blue is that this dye will stain viable cells over time in a cell-specific manner. It is therefore critical to optimize trypan blue protocols for each cell type and experiment.
All of these dyes can be used on single cells and tissues and evaluated using fluorescence microscopy or flow cytometry, although trypan blue staining is more commonly evaluated by counting cells manually using a hemocytometer and microscope.
Unlike cell impermeable nucleic acid stains, cell permeable nucleic acid stains, such as acridine orange, readily diffuse across cell membranes and thus stain the DNA of viable cells. Nonviable cells are not efficiently stained as their compromised membranes let the dye easily diffuse out of them. Acridine orange is a dual-fluorescence stain that emits green fluorescence when bound to dsDNA and red fluorescence when bound to ssDNA and RNA. When using acridine orange as a DNA binding dye, keep in mind that it does not efficiently bind condensed chromatin. As discussed in section 6, this dye can also be used to assess the ability of cells to maintain acidic pH in lysosomes, another indicator of cell viability. Cell permeable nucleic acid stains can be used to identify and quantify viable cell populations by microscopy, flow cytometry, or microplate assay.
Cell impermeable and permeable dyes can be used in combination with fluorescently labeled antibodies against proteins of interest, making it simple to include crucial viability controls in every experiment. A key consideration when using these dyes to assess membrane integrity is that different cell types have different membrane-permeability characteristics. For example, some cell impermeable DNA stains will readily stain dead eukaryotic cells but are unable to stain prokaryotes with compromised membranes.
Membrane integrity can also be assessed using enzymatic assays. A popular choice is assessing leakage of the cytoplasmic enzyme lactate dehydrogenase (LDH) into the culture medium on cell death. LDH colorimetric assays can be carried out in microplate format and read using an ELISA microplate reader.
Membrane disintegration can also be measured using chromium release assays. Here cells are pre-labeled with 51Cr prior to experimental treatment. Chromium binds to cellular proteins and is released into the culture medium upon cell death. By determining the amount of free versus bound 51Cr, using a scintillation counter, cell death can be measured quantitatively. Cells can also be prelabeled with fluorescent dyes. These methods sometimes suffer from high background because of spontaneous release of labels; they are useful only for applications that permit prelabeling of cells of interest.
2. Enzymatic activity
Another key distinction between viable and nonviable cells is that viable cells are enzymatically active. Esterase activity, as measured using substrates such as carboxyfluorescein diacetate (CFDA), is commonly used as an indicator of overall enzymatic activity. CFDA is a nonfluorescent, cell permeable dye that is hydrolyzed to form the fluorescent molecule carboxyfluorescein by nonspecific intracellular esterases present only in viable cells. Because carboxyfluorescein is negatively charged, it is retained noncovalently in viable cells, resulting in green fluorescence.
CFDA-SE is a derivative of CFDA that is better retained upon hydrolysis. Its product, CFSE, has succinimidyl ester groups that form covalent bonds with primary amines of intracellular proteins, allowing long-term tracking of cell viability. Because CFSE is covalently retained in cells, it is a more reliable indicator of cell viability and a popular measure of cell proliferation, as its fluorescence is halved with each cell division (Figure 3). Improvements to CFDA-SE have been made with proliferation dyes that have greater cell retention, provide greater resolution of cell division, and are available in a wide range of excitation and emission spectra to enable incorporation into multicolor fluorescent applications such as flow cytometry.
A.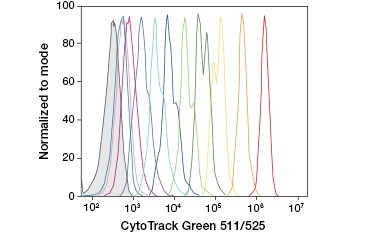
B.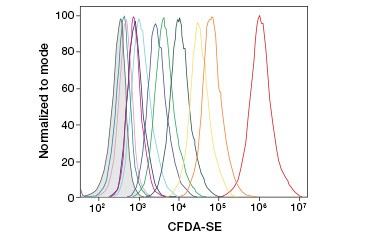
Fig. 3. Tracking of cell generations by flow cytometry. Ten cell generations can be resolved using CytoTrack™ Green 511/525 Cell Proliferation Assay (A) and up to eight using CFDA-SE (B).
Many other fluorescent esterase substrates, such as calcein derivatives and BCECF, are available with varying properties, including differences in excitation/emission spectra. Because BCECF’s fluorescence is pH sensitive, it can also track changes in cytosolic pH.
Using cell permeable esterase substrates, viability can be assessed by fluorescence microscopy and flow cytometry. These substrates can also be applied to tissue samples or used in high-throughput microplate assays.
3. Metabolic activity
Another common measure of cell viability is metabolic activity, which can be evaluated using oxidizable and reducible probes.
Resazurin is a popular oxidation-reduction (redox) indicator. This cell permeable dye can be added directly to cell culture media, making it a fast and convenient assay. In metabolically active cells, blue and weakly fluorescent resazurin is reduced to form resorufin, a pink and highly fluorescent molecule. This change can be monitored using both fluorescent and colorimetric assays. Because the amount of fluorescence is directly proportional to the number of living, respiring cells, resazurin is widely used to quantitatively measure cell viability. Resazurin is a convenient, more sensitive and stable, and non-toxic alternative to other redox indicators such as the tetrazolium salts MTT and XTT. Unlike tetrazolium salt-based assays, which are colorimetric and can only be read by using a microplate reader, resazurin viability assays can be read using flow cytometry or fluorescence microscopy. Resazurin and tetrazolium salts are particularly useful for high throughput applications because viability can be assessed in microplates using a microplate reader (Figure 4). XTT is often chosen over MTT for higher throughput applications because unlike MTT it yields a water soluble dye, thus eliminating a solubilization step that is crucial for MTT assays. Resazurin and tetrazolium salts are most commonly used to assess viability of adherent cells and cells in solution.
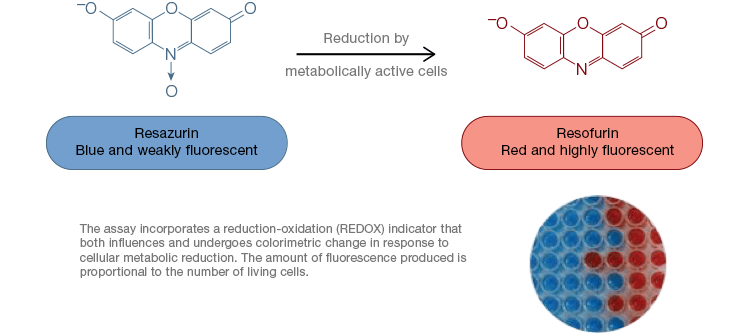
Fig. 4. Nontoxic resazurin oxidation assays are easily scalable for high-throughput applications. Oxidation of alamarBlue in metabolically active cells results in a colorimetric change from blue to red as well as an increase in fluorescence.
Dihydrorhodamines, -calceins, and -fluoresceins are reduced-rhodamine, -calcein, and -fluorescein derivatives that are cell-permeant and colorless. These compounds are readily oxidized to their fluorescent parent dye by reactive oxygen species present in living cells and tissues.
Another class of viability assays that measure metabolic activity are luminescent ATP assays. These quantify ATP released by cells upon lysis. They do not require long incubation periods, are extremely sensitive, and are commonly read using a microplate reader.
One factor to consider when using assays that measure metabolic activity is that metabolic activity can differ under different cell culture conditions. Proper controls thus need to be included to account for these differences.
4. DNA synthesis/replication
Another measure of viability, and a popular method of assessing proliferation, is quantification of BrdU incorporation into newly synthesized DNA. BrdU-containing DNA is identified using conjugated BrdU antibodies or BrdU antibodies in combination with a conjugated secondary antibody (Figure 5). These assays allow a variety of readouts as antibodies can be coupled to different types of indicators (fluorophores, peroxidases, etc.). Antibody labeling of specific cell markers also allows specific proliferating populations to be distinguished. Samples are fixed post-incubation and then analyzed using flow cytometry, fluorescence microscopy, immunohistochemistry, or microplate readers. Using flow cytometry, cells can be attributed to the G0/G1, S, or G2/M phase of the cell cycle based on their total DNA content. Using chromogenic peroxidase substrates allows assays to be read without a fluorometer. And by using [3H]-TdR instead of BrdU these assays can be converted to a radiolabel readout.
A.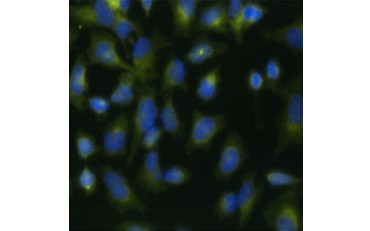
B.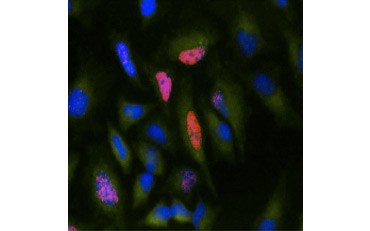
Fig. 5. Visualization of BrdU incorporation. HeLa cells were treated with 10 µg BrdU for 1 hr (B) or left untreated (A). Cells were stained with Mouse Anti-BrdU Antibody, clone Bu20a (MCA2483; 1:25 dilution) and Goat Anti-Mouse IgG (H/L) DyLight 549 Conjugated Antibody (red) (STAR117D549GA; 1:50 dilution) was used as a secondary antibody. The cytoplasm was stained with Rabbit Anti-GAPDH Antibody (AHP1628; 1:100 dilution) and Sheep Anti-Rabbit IgG DyLight 488 Conjugated Antibody (green) (STAR36D488GA; 1:50 dilution) was used as a secondary antibody. PureBlu™ DAPI (1351303) was used as a nuclear counterstain.
5. Proliferation marker expression
Certain proteins are known to increase in expression during cell proliferation. Ki-67, for example, is expressed in the nucleus of dividing cells but is not detectable during G0 and G1 (Figure 6). Similarly, histone H3 phosphorylation can be used to identify cells in M phase. Other commonly used proliferation markers include proliferating cell nuclear antigen (PCNA) and minichromosome maintenance 2 (MCM2). Using antibodies against these, other key proliferation markers or cell lineage markers can be assessed using ELISA, flow cytometry, immunohistochemistry, fluorescence microscopy, and Western blotting.
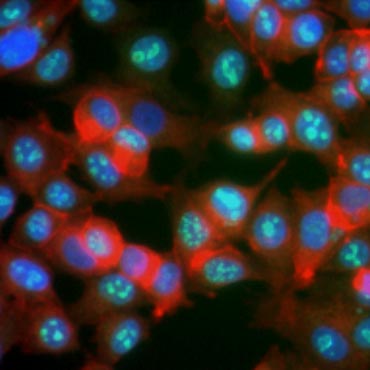
Fig. 6. Antibodies against Ki-67 identify proliferating cells. Staining of MCF-7 cells with human anti Ki-67 (HCA006, green) counterstained with phalloidin (red) and Hoechst (blue).
6. Membrane potential and pH
Live cells actively maintain ion gradients across their membranes, generating transmembrane potentials. When a cell dies these ion gradients are disrupted. This can be assessed using potentiometric dyes. JC-1 is a commonly used potentiometric dye. In healthy cells JC-1 localizes to the mitochondria, where it forms red fluorescent aggregates. Upon breakdown of the mitochondrial membrane potential, JC-1 diffuses throughout the cell and exists as a green fluorescent monomer. Tetramethylrhodamine ethyl esters (TMRE) and Tetramethylrhodamine methyl esters (TMRM) like JC-1 localize to mitochondria in healthy cells and to the cytoplasm in dying cells. Unlike JC-1 these dyes do not display a shift in their emission spectra. Instead, mitochondrial outer membrane permeabilization results in a decrease in TMRM/TMRE red fluorescence.
Viable cells are able to generate and maintain low pH in the cell’s acidic organelles, such as the lysosome. This can be measured using acidotropic stains such as acridine orange and neutral red. One consideration when using acridine orange is that this dye also binds DNA and RNA. When bound to single strand nucleotides, acridine orange fluoresces red; when bound to double strand DNA, it fluoresces green; and when bound to DNA in the lysosome, it fluoresces orange.
Regardless of the method chosen, it is critically important to include appropriate controls when performing any of the assays described here, as experimental conditions and cell type can alter the performance of some assays. Always include positive and negative controls. When using probes in solvents, also include vehicle-treated controls to account for effects of the solvent. Use application specific controls to control for factors such as autofluorescence in immunofluorescence and flow cytometry experiments and loading controls for Western blotting.
For information about Bio-Rad’s cell viability and proliferation assays, visit our cell health range page.
Read Part II Assessing Cell Health: ApoptosisalamarBlue is a trademark of Trek Diagnostic Systems. DyLight is a trademark of Thermo Fisher Scientific.


