We are all born with disease hardwired into our genome. A few mutations paired with lifestyle choices or other environmental factors can lead to asthma, Alzheimer’s, and thousands of other conditions. One out of every 100 of us suffers from a disease caused by a mutation in just a single gene. A particular group of drugs, antisense and gene therapies, tackle these disorders at their origin, the faulty gene or its expression. But even the most successful of these genetic therapies provide only a treatment. A new approach, gene editing, now promises more – a cure. Using tools like zinc finger nucleases (ZFNs), TALENs, and CRISPR scientists and doctors are erasing disease-causing mutations, providing a permanent cure and changing the way we approach and think about healthcare.
Traditional genetic therapies: knocking in or knocking down
The idea that “good DNA” could replace faulty DNA to cure disease is almost 50 years old and for decades gene and antisense therapies have shown the greatest potential for turning the dream of genetic therapy into reality.
Gene therapy combats faulty DNA by providing a transgene. This foreign gene is semi-randomly inserted into the patient’s genome, usually using a virus, conferring a desired new function to target cell types or tissues. One class of gene therapies, called gene augmentation, can be used to resolve recessive loss-of-function disorders, like cystic fibrosis, where a single wild-type gene is sufficient to restore health (Figure 1A). Gene inhibition therapies, on the other hand, provide a transgene that mitigates the effect of a toxic gain-of-function mutation (Figure 1B). This approach is particularly promising for cancer treatments where it can be used to counteract the activity of oncogenes. A third approach uses transgenes as suicide genes that kill target cells or as molecular beacons that mark specific cells for destruction (Figure 1C).
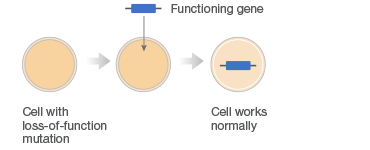
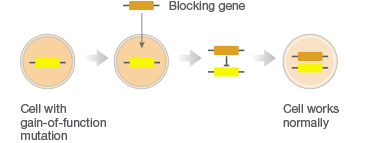
C. Killing of Diseased Cells
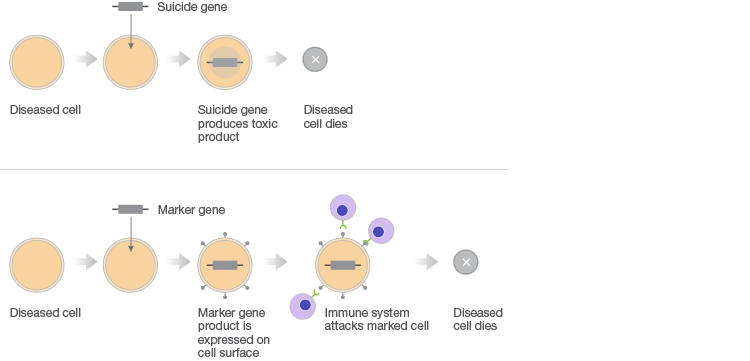
Fig. 1. Different gene therapy approaches.
Antisense therapies are similar to gene inhibition therapy but instead of providing “good DNA,” antisense therapies interfere with the expression of “bad DNA” at the mRNA level (Figure 2). Drugs like antisense oligonucleotides (ASOs) and small interfering RNAs (siRNAs) mitigate the presence of a disease-causing mutation by either preventing translation of target mRNAs, causing mRNA degradation, or by altering how mRNAs are assembled during splicing.
Antisense Therapy
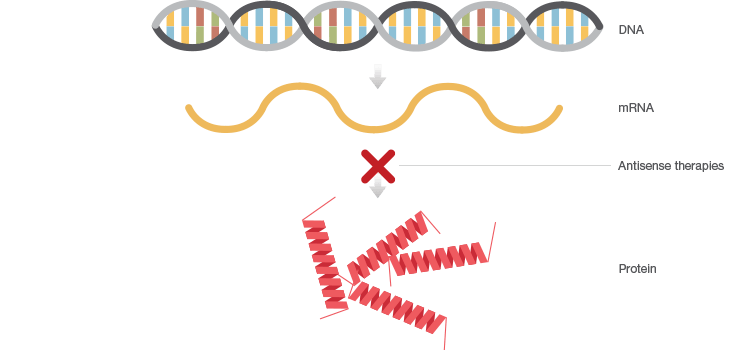
Fig. 2. Antisense therapies interfere with the expression of faulty DNA by preventing translation of corresponding mRNA.
A treatment not a cure
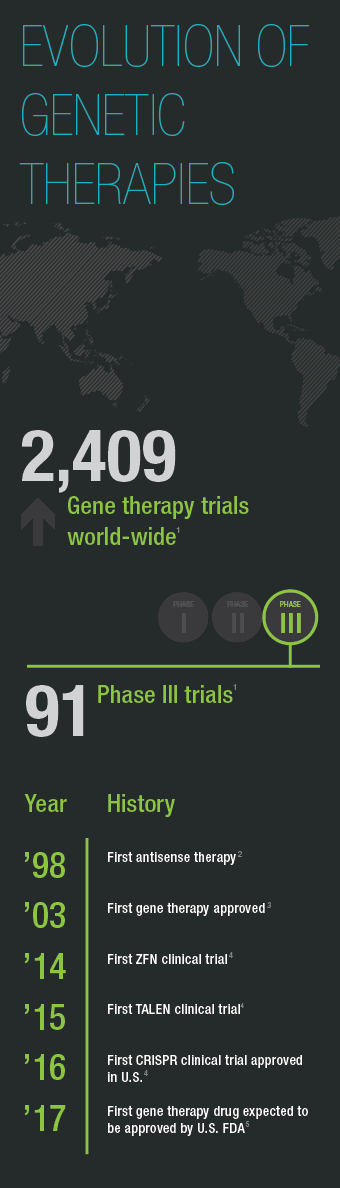
- Wiley Database on Gene Therapy Trials Worldwide, http://www.abedia.com/wiley/index.html, accessed March 17, 2017.
- Lundin KE et al. (2015). Oligonucleotide therapies: The past and the present. Hum Gene Ther 26, 475–485.
- Wirth T et al. (2013). History of gene therapy. Gene 525, 162–169.
- Reardon S. (2016). First CRISPR clinical trial gets green light from US panel. http://www.nature.com/news/first-crispr-clinical-trial-gets-green-light-from-us-panel-1.20137, accessed March 7, 2017.
- Mullin E. (2016). Gene therapy in U.S. is on track for approval as early as next year. https://www.technologyreview.com/s/602625/gene-therapy-in-us-is-on-track-for-approval-as-early-as-next-year/, accessed March 7, 2017.
Both antisense and gene therapy approaches took decades and many clinical trials to make their way to market. The first gene therapy, Gendicine, was approved in 2003 in China, more than 13 years after the first gene therapy clinical trial suggested that gene augmentation could be used to treat disease. It delivers a functional copy of the p53 gene for the treatment certain cancers (Pearson 2004). To date, no gene therapy is approved for use in the United States, but that is expected to change this year. GlaxoSmithKline’s gene augmentation therapy for a severe immune disorder called ADA-SCID and Spark Therapeutic’s treatment for a form of progressive blindness are vying to become the first FDA-approved gene therapy. Antisense drugs, on the other hand, are already available in the U.S. including treatments for familial hypercholesterolemia and spinal muscular atrophy.
Despite these successes, gene and antisense therapy share a significant caveat – they leave the faulty gene intact. In the case of gene therapy this means that treatment is only in place as long as the transgene remains integrated into the patient’s genome. In the case of ASOs and siRNAs, treatment is even more transient, as it is limited by the half-life of the short oligonucleotides used for treatment. Gene and antisense therapy thus rely on the continued presence of the therapeutic transgene or oligonucleotide. For diseases like cancer, treatment has to be in place until the disease is gone. For genetic disorders, like Duchenne’s or cystic fibrosis, treatment is required for the patient’s entire lifetime. Gene editing is the new approach that promises to do away with these long-term efficacy concerns by overwriting mistakes in the genome.
Erasing disease-causing mutations
Instead of relying on a transgene or temporary knock-down of gene expression, targetable endonucleases like ZFNs, TALENs, or CRISPR can be used to correct mutations and effectively erase them from a patient’s genome. This drastically shifts our approach allowing us to move from disease management or treatment to restoring wild-type phenotypes and providing a true cure.
In addition, because gene editing technology can be used to target any piece of DNA in a patient’s genome it is not limited to treating diseases caused by mutations in protein encoding genes. This greatly expands the number of diseases that can be treated. For example, we can now target mutations in regulatory elements such as promoters, noncoding RNAs, and miRNAs that are associated with diseases like Alzheimer’s, schizophrenia, and hearing loss.
To clinical trials in record time
Another benefit of gene editing is its relative ease of use which has allowed this new genetic therapy approach to move into and through clinical trials at record speed. Some of the most promising trials have used gene editing not to correct mutations but for cell based therapies that confer protective or disease-fighting features to patient-derived or donor cells. Cells are edited ex vivo and then transfused into a patient to combat disease. This allows more efficient gene editing than using current technology to target specific cell types in vivo.
One such therapy is a Sangamo Therapeutics ZFN therapy for HIV that introduces a naturally occurring mutation into T-cells that seems to confer resistance to infection with the most common HIV strain. This drug is now in Phase 2 clinical trials with the preliminary results that two out of three patients have remained off antiretroviral therapy for over one year (Sangamo Therapeutics 2015).
A trial at London’s Great Ormond Street Hospital also used edited T-cells but in this case scientists took advantage of gene editing’s ease of use to design cells with multiple altered genes. A one-year old girl with terminal acute lymphoblastic leukemia has been in remission after transfusion with donor T-cells in 2015 that were edited using TALENs to (1) seek out and destroy cancerous cells and (2) modify genes that protect the new T-cells from anti-cancer drugs. This therapy, UCART19, developed by Cellectis is now in Phase I clinical trials (Zusi 2015; Cellectis 2016).
Although ZFNs and TALENs have been used successfully in clinical trials, the standout gene editing tool is CRISPR. This technology has been available as an editing tool for less than six years yet clinical trials are already underway. One, at Sichuan University’s West China Hospital, uses CRISPR-edited T-cells to treat patients with advanced lung cancer (Cyranoski 2016). In the U.S. an NIH advisory committee approved the first CRISPR clinical trial in 2016 and CRISPR startup Editas plans to begin its first clinical studies in mid-2017 for in vivo gene editing to correct a mutation that results in blindness (Reardon 2016).
The seemingly limitless promise of CRISPR
Fueled by these rapid and early successes, a growing number of startups are developing CRISPR-based therapies, many backed by multi-hundred million dollar investments from large pharmaceutical companies like Novartis, AstraZeneca, and Pfizer. Meanwhile, translational research in academic laboratories is generating proof-of-concept data at breakneck speed for treatments of diseases that range from Duchenne muscular dystrophy and Parkinson’s to ZIKA and malaria.
Like the development of gene and antisense therapies, CRISPR therapies face hurdles. Off-target edits, for example, are of concern, especially given the permanent nature of gene editing. But part of the excitement surrounding CRISPR is how quickly these hurdles are cleared. Already multiple variants of the CRISPR system have been created that reduce off-target editing and CRISPR variants including inducible versions or ones that modulate gene expression without changing DNA sequence have been developed. The rate at which the CRISPR toolkit is expanding and being adapted to meet existing and new needs fuels the excitement and hope surrounding this new technology. SCID gene therapy took 26 years to make its way to market and still requires approval for use in the United States. CRISPR gene editing, on the other hand, has moved from laboratory to clinical trial in less than six years.
Reinventing the meaning of disease
It is not just CRISPR technology that is evolving at an unprecedented pace, it is also our attitude towards gene editing. Only a year ago scientists called for a moratorium on human germ line editing. Altering germ line cells with mutations that would be passed on from generation to generation was unthinkable. Last month, however, the National Academy of Sciences (NAS) reversed its stance on human germ line editing. In a joint report with the National Academy of Medicine, the NAS announced that germ line editing is permissible if certain conditions are met. In the absence of reasonable alternatives, germ line editing of genes that have been “convincingly demonstrated to cause or predispose” a patient to a serious disease may be permissible (NAS and NAM 2017).
This signals a tremendous shift in how we approach the ethics of gene editing and envision the future of genetic therapies and healthcare. In only a few years we have moved from viewing any type of human gene editing as worrisome science fiction to beginning to consider even its most permanent form, germ line editing, as a potential part of our treatment repertoire. Even though technical challenges currently prevent us from safely editing human germ line cells it is not difficult to imagine a future with a fundamentally different concept of disease. A future where gene editing can be used to erase disease-causing mutations and eradicate genetic-based disease.
References
Cellectis (2016). Cellectis announces first patient treated in Phase 1 trial of UCART19 in pediatric acute B lymphoblastic leukemia (B-ALL). https://www.cellectis.com/en/press/cellectis-announces-first-patient-treated-in-phase-1-trial-of-ucart19-in-pediatric-acute-b-lymphoblastic-leukemia-b-all, accessed January 24, 2018.
Cyranoski D (2016). CRISPR gene-editing tested in a person for the first time. Nature 539, 479.
Mullin E. (2016). Gene therapy in U.S. is on track for approval as early as next year. https://www.technologyreview.com/s/602625/gene-therapy-in-us-is-on-track-for-approval-as-early-as-next-year/, accessed March 7, 2017.
NAS and NAM (2017). Human genome editing: Science, ethics and governance. http://nationalacademies.org/cs/groups/genesite/documents/webpage/gene_177260.pdf, accessed January 24, 2018.
Pearson S et al. (2004). China approves first gene therapy. Nat Biotechnol 22, 3–4.
Reardon S. (2016). First CRISPR clinical trial gets green light from US panel. http://www.nature.com/news/first-crispr-clinical-trial-gets-green-light-from-us-panel-1.20137, accessed March 7, 2017.
Sangamo Therapeutics. (2015). Sangamo BioSciences presents Phase 2 clinical data from two SB-728-T HIV studies. http://investor.sangamo.com/press-releases/detail/302/sangamo-biosciences-presents-phase-2-clinical-data-from-two, accessed March 7, 2017.
Zusi K (2015). Gene editing treats leukemia. http://www.the-scientist.com/?articles.view/articleNo/44431/title/Gene-Editing-Treats-Leukemia/, accessed March 7, 2017.
Gendicine is a trademark of Shenzhen SiBiono GeneTech.