Is the hype surrounding biosimilars real or simply a hopeful view on an exceedingly expensive drug market? Substantial savings with these effective alternative treatments have been promised, but can they really deliver a viable alternative treatment?
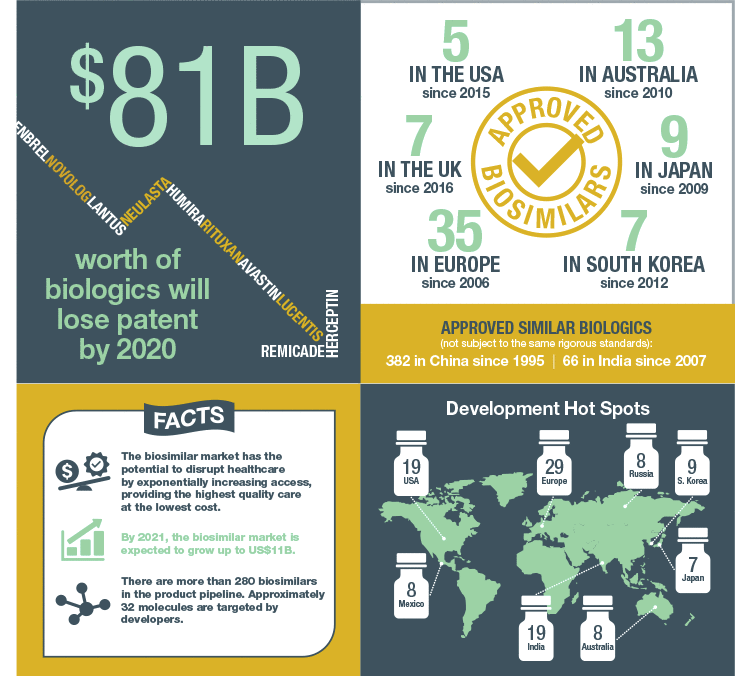
The biosimilars market is expected to grow to US$11 billion by 2021 from US$3.4 billion in 2016. Approval of monoclonal antibody biosimilars, better clarity in biosimilar regulations in major markets, and growth of sales in emerging markets are at the heart of this rapid expansion (Challener 2014). So what exactly is a biosimilar and why do these therapeutics have the potential to take over the biopharmaceutical market?
What are Biosimilars?
A biosimilar is “a product that is highly similar to the reference product notwithstanding minor differences in clinically inactive components; and there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product,” said Emanuela Lacana, PhD, Associate Director, Biosimilars and Biologics Policy, Office of Biological Products, CDER, FDA, at the PDA/FDA Joint Regulatory Conference in Washington, DC in September 2016 (Tierney 2016). More simply, it is a biologic that is almost identical to a previously approved biological product, with no clinically meaningful differences in safety or efficacy.
Originator biologics have provided lifesaving treatment for cancer, inflammatory bowel disease, as well as several other diseases. However, the exorbitant cost of a biologic treatment regimen (up to $200,000 per year) is considered cost-prohibitive for many patients and a financial burden on insurance payers and national health agencies. In spite of these exorbitant costs, global sales of biologics are increasing rapidly and currently account for 20% of the global pharmaceutical market.

Why is the Cost of a Biologic so High?
The high price is driven in part by the considerable investment required by pharmaceutical companies, approximately $2.6 billion to develop a new biologic, and development time of over a decade (DiMasi et al. 2016). As modern health care expands into developing nations, governments and insurers are seeking less costly alternatives to bring affordable, life-saving biologic therapy to patients worldwide. In addition, the upcoming patent expirations for several bestselling biologics have incentivized many pharma and biotech companies to pursue development of biosimilars, which are less expensive to develop, but that also contain biologic properties similar to their reference drug. The European Union (EU) approved its first biosimilar in 2006 and leads the global biosimilar market with 35 approved products. In addition, they were the first to develop product-specific development guidelines.
However, uptake of biosimilars is slower than expected globally. Patients, prescribing physicians, and pharmacists are concerned about safety and efficacy, non-approved indications, and the lack of a standardized naming system among global regulatory bodies. In addition, insurance payers and pharmaceutical companies are concerned that the introduction of biosimilars into the pharmaceutical market is economically unpredictable and may negate the cost advantage.
How Similar Does a Biosimilar Need to Be to Its Reference Product?
Although the definition of a biosimilar varies, all regulatory bodies state that a biosimilar must be a biologic, have a biologic reference product, the so called originator that is licensed based on a non-abbreviated drug submission, and be highly similar to the reference product in terms of safety, quality, and efficacy (Choy and Jacobs 2014).
Unlike the development of a chemically identical generic, biologic manufacturing uses a unique cell line and a complex, proprietary manufacturing process that is impossible to replicate entirely. All biologics have microheterogeneity. Changes to the manufacturing process mean that even an originator biologic may not be identical across production batches. And so to ensure that quality, safety, and efficacy are unaffected, biologic products undergo physicochemical and functional comparability experiments before and after manufacturing modifications. Similarly, biosimilar development requires extensive physicochemical and biological characterization of the reference originator product using multiple analytical techniques to ensure similarity. Based on the results of this characterization, preclinical studies and clinical trials are undertaken to resolve uncertainty about the biosimilarity of the new biosimilar product.
Demonstrating Biosimilarity: Comparison of Efficacy, Safety, and Quality
Although regulatory processes for establishing biosimilarity vary among countries, all require the use of a single reference originator product for comparisons of efficacy, safety, and quality.
Preclinical Studies
In preclinical studies, comparison is required for the proposed vs. the reference originator product to examine protein structure, enzymatic posttranslational modifications, potential protein variations, and intentional chemical modifications in multiple representative lots. In vitro, in vivo, and functional assays should demonstrate that the proposed product has highly similar biologic activity and potency and no clinically meaningful differences from the reference product (Ventola 2013).
Clinical Trials
Authorities generally agree that data on human pharmacokinetic/pharmacodynamic (PK/PD) responses are fundamental for supporting biosimilarity and require a clinically relevant study population, dose, and route of administration.
The design of comparative clinical studies for biosimilars to investigate efficacy, safety, tolerability, and immunogenicity, vary based on the findings and limitations of previous testing, the extent to which human PK/PD data predict clinical outcomes, and the extent of clinical experience with the reference originator and proposed biosimilar. An equivalence design is usually used to confirm that the proposed biosimilar is neither inferior nor superior to its reference originator product. Assessment of immunogenicity is critical because it can alter the PK or promote development of antidrug antibodies, thereby affecting safety and efficacy of the proposed drug. Because the required sample size for clinical trials is smaller for biosimilars than for originator biologics, careful monitoring of immunogenicity and other side effects is important in post-marketing surveillance of biosimilars (U.S. Department of Health and Human Services, 2015).
Concerns about Biosimilars
As more biosimilars enter the development pipeline, the general nature of regulatory guidance documents has raised concerns about the extent of research required to confirm safety and efficacy, the desire to extrapolate results for unapproved indications, the standardization of naming, and the economic impact on the pharmaceutical market.
How Much Research Is Too Much?
Because biosimilar manufacturers do not have access to the documentation related to reference originator product development, the potential for differences between the biosimilar and the reference originator product increases after a manufacturing change in the latter. Over time, the differences in manufacturing and quality control of reference originator products and biosimilars could lead to clinically significant differences between the products. Thus, the FDA requires more data to establish biosimilarity than to establish comparable effects in a reference originator product.
Despite the implementation of guidelines or draft guidance among most authorities involved in biosimilar manufacturing, including the WHO, guidelines are still somewhat general and not standardized. A systematic review of the regulatory pathways for the first 21 biosimilars approved in the European Union (EU) showed high variability in the number and size of PK/PD and phase III trials, trial design, dosing, trial endpoints, equivalence margins, and statistical methods (Mielke et al. 2016). It was concluded that while most sponsors followed the regulatory guidelines for biosimilar development published by the European Medicines Agency (EMA), some obtained approval without strict adherence to the guidelines.
Overall, the studies are focused on resolving areas of residual uncertainty and assessing biosimilarity using a totality of evidence approach, without specific limits or upper outcomes. While flexibility in requirements is important, narrower guidelines directed toward specific types of biosimilars may help guide and standardize development within different classes of drugs. The EMA has published product-specific guidelines for multiple drug classes, including recombinant erythropoietins, somatotropin-containing products, monoclonal antibodies, recombinant insulin and insulin analogs, and granulocyte colony-stimulating factor. Many experts believe that targeted guidelines will help direct the research needed to prove biosimilarity while maintaining the flexibility needed to accommodate new types of products.
Safety of Extrapolation for Biosimilars
Many authorities, including the EMA and U.S. FDA, state that extrapolating a biosimilar for an indication approved in the reference originator product is acceptable if scientific evidence confirms that the biosimilar has the same mechanism of action, target-binding characteristics, pharmacokinetics, and biodistribution in the approved and extrapolated indications (Cauchi 2017). Although extrapolation of biosimilars has been shown to be safe in the EU, clinicians and professional societies (such as the National Comprehensive Cancer Network and American College of Rheumatology) express concern about the lack of rigorous data investigating long-term effects (Camacho et al. 2014). According to an editorial published by members of the Working Party on Similar Biological (Biosimilar) Medicinal Products, missing clinical trial data specific for the extrapolated indication may discourage clinicians from using the biosimilar (Weise et al. 2014). The authors suggested the need for additional data if the mechanisms of action and target receptor for the extrapolated indication are different or if the historical safety profile of the reference product differs between the two indications. Similarly, the National Comprehensive Cancer Network suggested developing specific recommendations for extrapolation in biosimilars and recommended against using biosimilars in populations for which insufficient data are available (for example, use of biosimilar granulocyte colony-stimulating factors in normal bone marrow donors) (Choy and Jacobs 2014).
What’s in a (Biosimilar) Name?
Standardizing a clear naming system for biosimilars has been hotly debated among different regulatory bodies as more biosimilars become approved. Most experts agree that standardized nomenclature helps minimize confusion with prescribing, dispensing, and performing post-marketing surveillance. However, regulatory authorities around the world diverge on whether biosimilars should have distinct names. To standardize the approach to identifying biosimilars, the WHO released a proposal in January 2016 for a worldwide naming convention that adds four random consonants and an optional two-digit checksum to the end of the international nonproprietary name (INN) of the original biologic (WHO, 2016). While the FDA’s guidance for biosimilar naming (a suffix of four random letters added to the core name of the reference drug) is similar to WHO’s recommendation, nomenclature convention still varies widely among other regulatory authorities. Japan requires the biosimilar to have the nonproprietary name of the reference product plus BS (for biosimilar) and a number that corresponds to the order in which it was approved, whereas South Korea does not require biosimilars to have a distinctive nonproprietary name. This lack of cohesion among regulatory authorities may make global tracking of the use, efficacy, and safety of the biosimilar more difficult.
Future Cost of Biosimilars
Although biosimilars are intended to relieve some of the cost burden, the economic effects of incorporating them into clinical practice are unclear. The wholesale price of Zarxio (filgrastim-sndz) was about 15% less than its reference product Neupogen (filgrastim) in the EU in 2009 and the U.S. in 2015. This modest price gap may cause slow uptake of biosimilars, particularly if physicians, patients, and insurance payers are reluctant to substitute the reference drug over lingering concerns of efficacy and safety. Furthermore, small price differences may incentivize the manufacturers of reference originator drugs to remain on the market, and the reference manufacturer may also lower the price of their drug to remain competitive. Alternatively, the price of a biosimilar may increase if the reference drug manufacturer chooses not to compete with the biosimilar. Taken together, the evolution of costs for biosimilars and reference drugs is unpredictable in these early stages and will likely determine the acceptance of biosimilars among patients, prescribing physicians, and insurance payers.
Future Implications for Biosimilars
Rapidly rising health care costs and impending patent expiration for top-selling biologics will continue to incentivize the inclusion of biosimilars into clinical practice. However, the actual effects of biosimilar development worldwide are still unclear. The uncertainty about legal issues (for example, patent infringement) and the complex expertise required to synthesize biologics will likely mean that biosimilar development will be limited to large pharmaceutical companies with secure finances and extensive experience. Furthermore, companies manufacturing reference originator products continue to investigate ways to offset the impending introduction of biosimilars by improving their first-generation product — reducing frequency of dosing or improving convenience of drug administration — which may extend patent protection or achieve new patent exclusivity, negating a company’s efforts in biosimilar development. Decreasing the cost of the reference originator product to narrow the price gap may be enough of a disincentive for prescribers and pharmacists prescribing a relatively unknown biosimilar instead of its rigorously tested reference counterpart.
Uptake of biosimilars has been slower than many experts anticipated, but in the 10 years since the first biosimilar was approved they have been shown to be safe and effective. Optimal inclusion of biosimilars as an alternate treatment will require that regulatory guidelines enable competition with reference products and increase confidence among patients, physicians, pharmacists, and insurance payers that biosimilars are safe and cost-effective. But to do so, the biosimilar industry will have to continue to prove that they can consistently meet, if not exceed, the standards of the already established biotherapeutic market. The pace and integrity with which biosimilar developers meet this challenge will eventually determine their fate.
References
Camacho LH et al. (2014). Biosimilars 101: considerations for U.S. oncologists in clinical practice. Cancer Med 3, 889–899.
Cauchi R (2017). State laws and legislation related to biologic medications and substitution of biosimilars. ncsl.org/research/health/state-laws-and-legislation-related-to-biologic-medications-and-substitution-of-biosimilars.aspx, accessed June 26, 2017.
Challener CA (2014). Focus on biosimilars. pharmtech.com/focus-biosimilars, accessed June 26, 2017.
Choy E and Jacobs IA (2014). Biosimilar safety considerations in clinical practice. Semin Oncol 41 Suppl 1, S3–14.
DiMasi JA et al. (2016). Innovation in the pharmaceutical industry: New estimates of R&D costs. J Health Econ 47, 20–33.
Mielke J et al. (2016). Clinical trials for authorized biosimilars in the European Union: a systematic review. Br J Clin Pharmacol 82, 1,444–1,457.
Tierney L (2016) Live from PDA/FDA: an overview of FDA’s expectations of biosimilars. healthcarepackaging.com/trends-and-issues/regulatory/live-pdafda-overview-fdas-expectations-biosimilars, accessed June 26, 2017.
United States Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research (2015). Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for industry. www.fda.gov/downloads/drugsguidancecomplianceregulatoryinformation/guidances/ucm291128.pdf, accessed June 26, 2017.
Ventola CL (2013). Biosimilars: part 2: potential concerns and challenges for p&t committees. P T 38, 329–335.
Weise M et al. (2014). Biosimilars: the science of extrapolation. Blood 20, 3,191–3,196.
World Health Organization. WHO informal consultation on international nonproprietary names (INN) policy for biosimilar products. who.int/medicines/services/inn/BiosimilarsINN_Report.pdf, accessed June 26, 2017.
Avastin, Herceptin, and Lucentis are trademarks of Genentech, Inc. Enbrel is a trademark of Immunex Corporation. Humira is a trademark of Abbvie Biotechnology Ltd. Lantus is a trademark of Sanofi-Aventis GmbH. Neulasta is a trademark of Amgen Inc. Novolog is a trademark of Novo Nordisk. Remicade is a trademark of Janssen Biotech, Inc. Rituxan is a trademark of Biogen MA Inc.