Researchers who use antibodies have a love-hate relationship with them. While we acknowledge that they are one of the most powerful tools at our disposal for the evaluation of proteins, our experiences with them have not always been pleasant or rewarding. However, the field of antibody technology has seen tremendous advances and, with all the recent developments and newly available techniques, this field has now come of age. The remarkable progress in our understanding of antibodies themselves and the availability of a vast array of advanced bioengineering tools have enabled us to produce extremely sophisticated antibodies with potential unimaginable until now. Maybe the nature of our relationship with antibodies is poised to take a turn for the better?
The Arduous Journey of Antibody Generation
We’ve come a long way from using polyclonal antibodies, through developing antibodies from immortalized antibody-producing cells to engineering different parts of the antibodies and generating antibodies using an animal-free library with billions of antibody fragments to choose from. Though popular for a very long time, the classic hybridoma technique proposed by Köhler and Milstein in 1975 had its drawbacks. It is based on the generation of monoclonal antibodies by injecting immunogenic antigens into mice, extracting B cells from their spleens, and then fusing those B cells with immortalized myeloma cells. The process involves several screening and selection steps before the desired monoclonal antibodies are generated. In addition, it is an extremely complicated and laborious process and its usefulness is further restricted by the fact that the antibodies can be derived only from animal sources.
Utilizing animal sources for generation of antibodies poses several other problems in addition to the cost, labor, and time factors. The immune response of animals to different antigens is variable and for a lot of conserved antigens not enough response is elicited to make them viable for generating antibodies. The limited immunogenicity of mammals to such conserved antigens also reduces diversity in immune response. Toxicity of certain antigens could also prevent successful antibody generation in animals. In addition, the use of animal-derived antibodies as biotherapeutics is also known to cause adverse effects, such as cytokine release syndrome in patients during drug testing. When used in clinical trials on humans, murine-derived antibodies trigger the production of human anti-mouse antibodies or human anti-rat antibodies, which can interfere with therapy or cause immune-complex hypersensitivity.
Humanizing Antibodies on the Way
The first attempts to make more “human friendly” murine-derived therapeutic antibodies involved creating chimeric versions of the antibodies by combining the variable binding domain of a mouse antibody with the constant domain of a human antibody (Morrison et al. 1984; Boulianne et al. 1984). But chimeric antibodies still exhibited some level of immunogenicity (Bruggemann et al. 1989), emphasizing the need for fully human antibodies for therapeutic uses. Also, there are limits to making animal-derived antibodies fully human. In fact, a 2010 review of publicly available data suggested that the alleged benefits offered by humanization are not that straightforward and must be evaluated on a case-by-case basis (Getts et al. 2010).
Finding a New Display Mechanism — the Phage Display Technology
The advent of recombinant antibody technology in the 1980s helped circumvent some of these problems by enabling researchers to isolate and express antibody genes using bacteriophage and cloning vectors. The phage display technique took this one step further by developing the ability to screen for a desired antibody by expressing the selected proteins on the surface of the phage. The technology involves fusing the selected proteins of interest with the endogenous phage coat proteins, producing fused hybrid proteins. A combinatorial library of the antigen binding fragments (Fabs) of the antibody producing cells, constructed using naïve or immunized B cells, can be incorporated into phage particles so that they are displayed on the surface of the phage. This library can then be screened against the antigen, which can be immobilized in the best functional conformation to ensure successful binding and selection. The phages that display antigen binding are then selected for large-scale amplification and enrichment to create antigen-binding clones in E. coli. After several such panning cycles, the library would be enriched for individual antigen-binding clones that can be isolated and expressed as monoclonal phages, and it would have specific antibodies that could be expressed recombinantly after testing and sequencing.
A.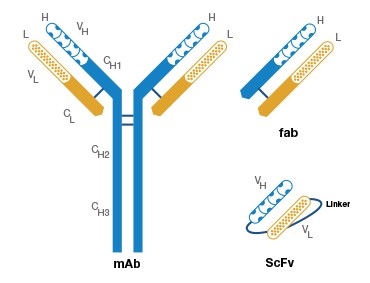
B.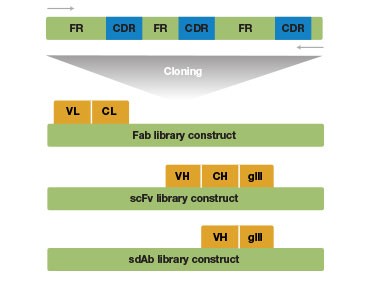
Fig. 1. Antigen binding fragments and complementarity determining regions (CDR) of an antibody. A, A Fab fragment consists of the variable domain and the corresponding first constant domain (VH–CH1 and VL–CL) in the antibody polypeptide chain. B, The CDRs are interspersed between the four conserved framework (FR) regions in the 5′ region of the antibody.
Incorporating foreign proteins into the phage host genome, however, has its own limitations. Some proteins cannot be expressed as hybrid fusions, and genomic incorporation of the proteins is also believed to impair the structural stability of the phage, resulting in reduced efficiency in its infectivity and propagation. Later attempts to develop phage display systems incorporated different coat genes for fusion or introduced a substitute copy of unmodified phage coat proteins to enhance binding affinity and to promote multivalent display. But the transformation efficiency of large phage vectors is low, and using the phage display system also imposes limitations on the isolation of higher affinity clones.
Circumventing Transformation Issues — the Use of Phagemids
The use of phagemids circumvents some of the issues involving transformation efficiency owing to their small size and the flexibility they offer to fuse the heterologous proteins with an additional copy of the coat protein. This way the expression of the gene and the production of phage particles are carried out by two different mechanisms. The phagemids carry the gene for the protein of interest and also provide replication machinery for the viral ssDNA, a selection marker, and an assembly for the ssDNA to be packaged into phage particles. When coinfected with helper phages, which possess all the components required for assembly and replication but lack a functional packaging mechanism, phagemids complement the production of phagemid DNA displaying the phagemid-encoded fusion protein.
Arriving at Large-Scale Phage Display Libraries
While many display libraries using ribosomes (Lipovsek and Plückthun 2004), bacteria, and yeast (Mattheakis et al. 1994) have also been developed, phage display libraries are the most popular and improved method of antibody selection thus far. Earlier methods adopted the fusion of the antibody heavy- and light-chain variable domains (as a single polypeptide chain called single chain Fv fragments (scFv)) to the coat protein of the filamentous phage (see Figure 1). While the use of scFv enabled antibody display as well as antigen binding, the affinity displayed by the scFv fragments was not as high as desired.
A significant advance in antibody display was the ability to display Fab fragments directly on the surface of the phage (Hoogenboom et al. 1991). This method took advantage of the fact that the coat proteins are naturally exported into the bacterial periplasm and that antibody heavy- and light-chain expression can be directed to occur in the periplasm using a signal sequence. So the antibodies are displayed as heterodimeric Fab fragments by linking one of the chains to the phage coat protein with a signal sequence and directing the complementary chain to be secreted into the bacterial periplasm. Since assembly of the chains occurs in the periplasm, the antibody fragments are displayed on the surface. The ability to display Fab fragments using phagemids opened the door for efficient construction of combinatorial phage libraries with the ability to use different combinations of antibody fragments.
These phage display libraries are constructed by amplifying and cloning the antibody variable domain genes with the viral coat protein into a phage display vector, which can display the entire immune repertoire. Antibodies binding to any specific protein can be obtained using these phage display libraries. Libraries can be constructed with natural sources (after immunization) using in vivo recombinations or by using synthetically derived chemical sources (synthetic libraries).
Discovering Synthetic Antibody Libraries
The biggest advantage of synthetic libraries is that they afford diversity unparalleled by natural sources. An endless number of possible combinations can be built in a synthetic antibody library by incorporating synthetic oligonucleotides into the CDRs of antibodies within the vectors used in phage display. In addition, the conditions for antigen binding can be manipulated in vitro until the optimal conditions are achieved, thereby ensuring that during the selection process the best specificity, highest affinity, as well as increased stability, and reduced cross-reactivity are obtained. A simplified version of how synthetic antibody libraries are created is shown in Figure 2.
A.
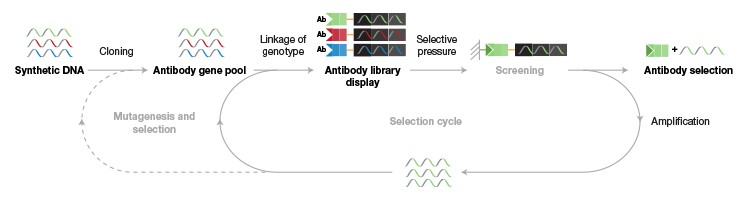
B.
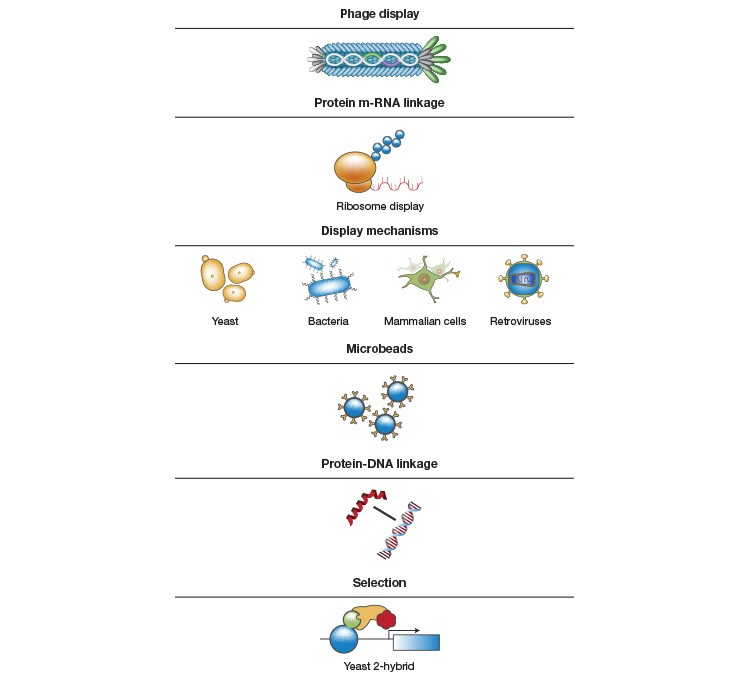
Fig. 2. Building synthetic antibody libraries. A, Synthetic oligonucleotides with various possible binding combinations and diversity are cloned into the vector used in phage display, thereby creating an antibody gene pool with diversity. To allow selection for antibodies with the desired characteristics, the specific genotype is linked to the antibody phenotype (for example, specificity and affinity), which can be used as a selection tool when selection pressure (for example, selecting for a chosen marker) is applied during the screening process. During the selection process, the phage is exposed to the antigens, and the specific phage carrying the antigen-binding Fab is selected in a biopanning process and propagated through E. coli to generate several antigen-binding clones. The process is repeated for several cycles until the library is sufficiently enriched for a particular clone. The individual clones can then be isolated and used for monoclonal antibody generation. B, Methods and tools that could be used for the various steps.
Arriving at a Juncture — the Transformative HuCAL Technology
The concept of a fully synthetic human combinatorial antibody library (HuCAL) was developed by Knappik et al. in 1999. It turned out to be a game changer in the way antibodies are generated. It is the first fully synthetic, fully human library ever developed. The HuCAL library was developed by an extensive analysis of the existing antibody diversity (the antibodies that are built during natural immune responses) and comparison to the germline repertoire (the antibodies that are present in human germline genes). It takes advantage of the fact that the antibody portions containing the antigen-binding Fv moiety share constant sequences that are perfectly superimposable upon each other, including upon sequences from other animal species. Accordingly, the library included consensus frameworks of seven VH and seven VL germline families, which were found to represent more than 95% of the human immune repertoire. This resulted in HuCAL master genes with a total of 49 combinations (seven for light chains and seven for heavy chains). The first library was constructed in the scFv format by diversifying the CDR3 regions (see Figure 1) with trinucleotide mixtures. Because these genes were synthesized artificially, the master genes could be made as modular as possible to get the best functional bodies with optimized expression and folding. The resultant library was thoroughly characterized by sequence analysis and by structural and functional analyses. It has also been automated for high-throughput antibody generation (Krebs et al. 2001).
The modularity of the HuCAL library offered the ability to convert it from the scFv format to other formats, in addition to the ability to exchange the CDR regions. As a consequence, the HuCAL-Fab 1 library was developed (Rauchenberger et al. 2003). Since the Fab fragments do not aggregate but remain as monomers (as opposed to scFv fragments, which tend to aggregate and form multimers), antibody screening could be accomplished based on affinity ranking and no purifications were required. This is a great advantage in terms of time savings during the antibody generation process. In addition, the Fab format can easily be converted into a complete immunoglobulin (IgG) format, which is ideal for therapeutic use.
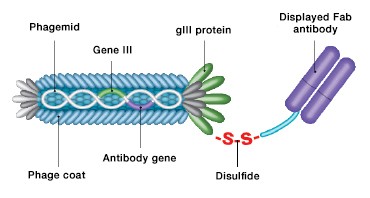
Fig. 3. CysDisplay screening technology. Fab fragments are linked to phage particles by a disulfide bond rather than a peptide bond. This allows elution of phage with reducing agents during antibody selection.
The Precious Advancements in the HuCAL Route
Further advancing HuCAL technology was the development of the HuCAL GOLD® (Rothe et al. 2008) and HuCAL PLATINUM® (Prassler et al. 2011) libraries. The HuCAL GOLD library was developed with diversification of all six CDRs, matching them to the sequences and variability of natural human antibodies from the corresponding germline families. This library also uses a novel display system called CysDisplay®, in which the antibody fragments are linked to the phage coat protein through a cleavable engineered intermolecular disulfide bridge and are displayed between the coat protein and the heavy chain of the antibody fragment (Figure 3). This facilitates the elution of specific phages by adding reducing agents rather than using affinity-dependent methods. The HuCAL GOLD library comprises more than 10 billion functional Fab fragments.
The HuCAL PLATINUM library (Figure 4) further improved the HuCAL GOLD library by optimization at various levels, such as those of genes and sequence spaces, so as to increase expression levels and avoid undesirable glycosylation sites. The diversification strategy was also improved to replicate the natural amino acid distribution, especially in certain CDR regions (HCDR-3). The HuCAL PLATINUM library facilitates isolation of fourfold more unique sequences than the GOLD version, and the full-length IgGs derived from the library are superior to those derived from the HuCAL GOLD library.
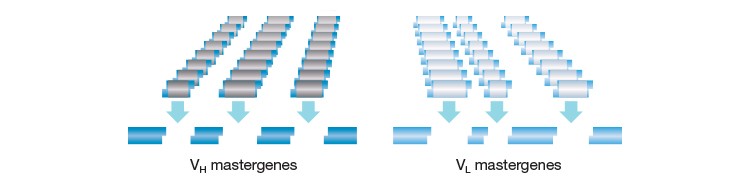
Fig. 4. The HuCAL concept. The structural diversity of the human antibody repertoire is represented by seven heavy chain and six light chain variable region genes, which are combined to produce 42 antibody frameworks in the master library. Superimposing highly variable genetic CDR cassettes on these frameworks effectively mimics the entire human antibody repertoire.
Realizing the Potential of the HuCAL Destination
HuCAL technology has since been commercialized, paving the way for rapid custom antibody generation and screening, and it has gained enormous popularity. Because of the vast antibody repertoire of the library, custom antibody generation and screening can be accomplished at mindboggling speeds (as little as eight weeks). The rapid process consists of selection, subcloning, screening, and production (Figure 5).
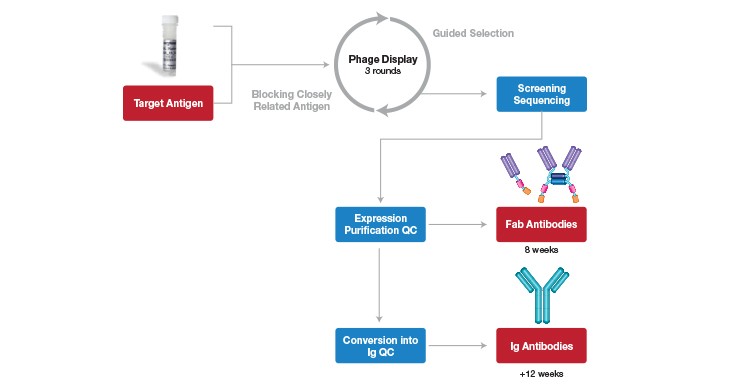
Fig. 5. HuCAL antibody generation and screening process. Target antigen immobilized on a solid support; guided selection for highly specific antibodies using 3 rounds of panning; screening of enriched specific antibodies on the target antigen and control antigens; sequencing of ELISA-positive clones; expression, purification and quality control of unique Fab antibodies (monovalent and bivalent formats); Fab antibodies can be generated within 8 weeks.
Ability to Select Antibodies with Remarkable Specificity
The in vitro generation of antibodies using the HuCAL technology facilitates extensive manipulation of antibody specificities, such that practically any desired specificity can be achieved. Different strategies can be established for selecting antibodies of particular interest. For example, using Guided Selection the conditions can be adjusted to select for specific epitopes, such as a phosphorylation site, an oxidation site, or a specific mutation. The antibody library is pre-adsorbed with the alternative state peptides (for example, a nonphosphorylated peptide or a wild-type peptide) to remove the antibodies that would bind to them, and the remaining phages are then used for selection against those peptides with the desired state. Pre-adsorption of the library is also advantageous in removing cross-reacting antibodies. Specific screening techniques can also be adopted for selecting antibodies that bind to homologous proteins (or different isoforms of a protein) or those that bind to antigens under specific conditions.
Ability to Rank Antibodies Based on Binding Strength
With an industrial production system, and given the ease with which monovalent Fabs can be generated using HuCAL, it’s feasible to add screening steps that allow selection for antibodies with a desired binding strength. Specific knowledge of binding strength is extremely useful for antibodies generated as reagents for use in the development of bioanalytical assays for therapeutic proteins. For example, FDA guidelines recommend inclusion of several antibodies with varying binding strengths as controls for the development of anti-drug-antibody (ADA) assays for immunogenicity testing. With custom generation of antibodies using HuCAL technology , high-throughput ranking of antibodies based on their koff rates can be accomplished rapidly (Ylera et al. 2013), enabling efficient antibody selection for assay development. The screening for koff rates doesn’t add a significant amount of time during antibody generation, but it does provide significant data for the assay development process, especially for generating antibody reagents to support therapeutic protein development.
Ability to Generate Antibodies with Different Formats
While monovalent or bivalent Fab antibody formats are useful for various reasons, such as elimination of nonspecific bindings to Fc receptors, prevention of cytokine release in immune assays, increased antibody diffusion, and co-crystallization of target proteins with antibodies, full-length IgGs are useful in immunodiagnostic assays as they provide better sensitivity in ELISAs and can also be used as human calibrators and standards. Because of HuCAL’s modularity, conversion from Fab to full-size IgGs is easily carried out when the Fc region is required for specific applications. The VH and VL regions are cloned into vectors with the desired constant regions and subsequently cotransfected for expression in mammalian cells. The Fab antibodies can be converted into different fully human Ig isotypes, such as IgG1, 2, 3 and 4, IgA, IgE, or IgM, which can be used as standards for the evaluation of ADA responses.
The modularity of the library also allows for the generation of chimeric antibodies when in vivo validation is required in animal models for proof-of-concept studies aimed at future drug development. In these cases, the HuCAL Fab antibody fragments are combined with the murine or rat Fc regions, converting them to full-length chimeric human-mouse or human-rat antibodies (Figure 6).
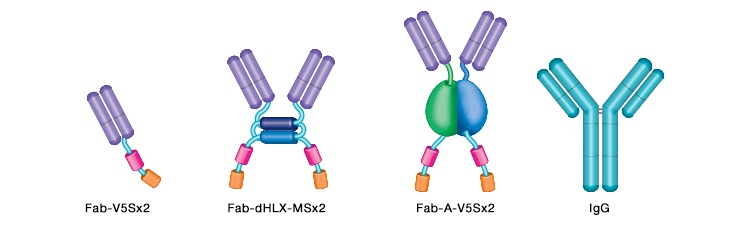
Fig. 6. Schematic of the main antibody formats available. From left to right: monovalent Fab shown with two tags; bivalent Fab, dHLX domains shown as blue cylinders; bivalent Fab formed by dimerization of bacterial AP (blue/green) with two tags; complete IgG antibody.
Ability To Be Used Directly for Immunogenicity Testing
Because the antibodies are developed entirely in vitro, and by virtue of their fully human nature, they can be used as positive controls for immunoassay standardization (Knappik et al. 2009). The technology also enables a high-affinity antibody to be used in pharmacokinetic studies as a Fab reagent first, and to then be converted into an IgG control for immunogenicity testing, thereby eliminating the use of patient or animal sera.
Tapping into the Potential of the HuCAL Technology and Continuing the Journey
Advances in science and the concurrent development of new tools provide opportunities for finding the answers to questions that might once have been unimaginable or would have taken a lifetime to answer. The development of the HuCAL technology is a significant milestone in enhancing the ability to develop specific antibodies as tools that can be used by the pharmaceutical and diagnostic industries and to unravel the functioning of specific proteins in academic labs. Embracing the technology can only advance such development and enable us to explore as yet untraversed territory.
The HuCAL technology is available as a custom antibody generation service from Bio-Rad, www.abdserotec.com/HuCAL.
References
Barbas CF 3rd et al. (1991). Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc Natl Acad Sci USA 88, 7,978.
Boulianne GL et al. (1984). Production of functional chimaeric mouse/human antibody. Nature 312, 643–646.
Bruggemann M et al. (1989). The immunogenicity of chimeric antibodies. J Exp Med 170, 2,153–2,157.
Chan CEZ et al. (2014). The role of phage display in therapeutic antibody discovery. Inter Immunol 26, 649–657.
Getts DR et al. (2010). Have we overestimated the benefit of human(ized) antibodies? mAbs, 2, 682–694.
Hoogenboom HR et al. (1991). Multi-subunit proteins on the surface of filamentous phage: methodologies for displaying antibody (Fab) heavy and light chains. Nucleic Acids Res 19, 4,133–4,137.
Knappik A et al. (2000). Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J Mol Biol 296, 57–86.
Knappik A et al. (2009). Development of recombinant human IgA for anticardiolipin antibodies assay standardization. Ann NY Acad Sci 1173, 190–198.
Köhler G and Milstein C (1975). Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497.
Krebs B et al. (2001). High-throughput generation and engineering of recombinant human antibodies. J Immunol Methods 254, 67–84.
Lipovsek D and Plückthun A (2004). In-vitro protein evolution by ribosome display and mRNA display. J Immnol Methods 290, 51–67.
Marks JD et al. (1991). By-passing immunization. J Mol Biol 222, 581–597.
Mattheakis LC et al. (1994). An in vitro polysome display system for identifying ligands from very large peptide libraries. Proc Natl Acad Sci USA 13, 9,022–9,026.
McCafferty J et al. (1990). Phage antibodies: filamentous phage displaying antibody variable domains. Nature 348, 552.
Morrison SL et al. (1984). Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci USA 81, 6,851–6,855.
Prassler J et al. (2011). HuCAL PLATINUM, a synthetic Fab library optimized for sequence diversity and superior performance in mammalian expression systems. J Mol Biol 413, 262–278.
Rauchenberger R et al (2003). Human combinatorial Fab library yielding specific and functional antibodies against the human fibroblast growth factor receptor 3. J Biol Chem 278, 38,194–38,205.
Rothe C et al. (2008). The human combinatorial antibody library HuCAL GOLD combines diversification of all six CDRs according to the natural immune system with a novel display method for efficient selection of high-affinity antibodies. J Mol Biol 376, 1,182–1,200.
Smith GP (1985). Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228, 1,315.
Ylera F et al. (2013). Off-rate screening for selection of high-affinity anti-drug antibodies. Anal Biochem 441, 208–213.


