It has been over 60 years since the first cytokine was identified. To date, more than 300 cytokines, chemokines, and growth factors have been studied with many functions involving the immune system (Turner et al. 2014). These cytokines often work in a network that modulates and regulates immune responses. Many of these cytokine molecules have an enigmatic role and in-depth research is essential to better understand the complexity of their association with various pathophysiological processes and the pleiotropic interactions that characterize the cytokine network.
Progress in mining these cytokine markers has shown that in drug discovery and the study of complex diseases, composite profiling of multiple biomarkers provides a more comprehensive understanding of drug effects and signatures in disease progression (Etzioni et al. 2003, Hsu et al. 2014, Williams 2009). The availability of commercial multiplex cytokine assays has greatly simplified this effort and enables end users to interrogate an entire network of cytokines in a single sample.
In small animal models, the ability to profile multiple cytokine markers using conventional single-plex ELISA techniques is often restricted by the requirement of a large sample volume. A typical 3-week-old mouse has a limited circulating blood volume of about 1.5–2.5 ml. Less than half of this volume is available in the form of plasma. This significantly limits the number of cytokine markers that can be studied per sample/animal. Consequently, studies that rely on only a few cytokines to determine whether they correlate with a physiological variable of interest may overlook the integrated effects of the complex cytokine network. In contrast, multiplex immunoassays offer a more comprehensive analysis whether for basic research, drug discovery, or monitoring of therapeutic interventions and provide considerable savings in both time and cost per assay.
Have Questions?
Ask UsWhile multiplex immunoassays continue to gain popularity in the fields of biomarker discovery and drug development, continuous improvement of these assays remains crucial in transitioning them to a mainstream role in research and eventually the clinical network. Here, we provide detailed insight into the SMARTS (Sensitivity, Multiplexibility, Actionable results, Reproducibility, Time, and Specificity) approach to better align user experience to the performance characteristics of these multiplex assays. The SMARTS concept has been implemented in product design, development, validation, and manufacturing of multiplex assays at Bio-Rad. The end goal of the SMARTS approach is to deliver end users with actionable results using the five performance characteristics of SMARTS to guide assay design and acceptance criteria.
The most pressing driver for adopting an improved immunoassay platform is greater assay sensitivity and the ability to measure low abundance proteins from complex sample matrices. For the Bio-Plex Multiplex Immunoassay System, we design and develop multiplex assays with sub-picogram per milliliter limit of detection (LOD) and a wide dynamic range (4 log) that enables detection of healthy, disease, and stimulated samples in the same assay. A wide dynamic assay range significantly increases the utility of a multiplex assay as a protein detection and quantitation tool in diverse autoimmune, inflammatory, cancer, infectious, and malignant conditions. Since the development and validation of multiplex assays are very complex and time consuming, we’ve used our vast expertise to develop novel approaches to address multiplex cross-talk, optimize reagents to improve sensitivity, apply a clinical-like approach to improve reproducibility, and develop antibodies and antigens for greater specificity. In today’s fast-moving world, researchers demand tools to expedite sample analysis and conserve resources. The SMARTS approach ensures end users gain new insights into their research and spend less time on the bench.

Antibody selection and reagent optimization
The ability to measure low abundance cytokines from complex sample matrices is essential to any immunoassay platform (Comley 2014). Measuring analytes well below the sub pg/ml range without adding complexity in amplification chemistry or extra steps to the standard assay workflow is the key to gaining acceptance among end users. Finding the best antibodies is vital to developing a sensitive assay. It’s best to use custom antibodies that are developed and screened for multiplex immunoassay applications. Many commercially available antibodies are suitable for traditional ELISA assays or western blotting but lack sensitivity and specificity in multiplex format. Antibodies prepared by positive affinity purification and fractionation by ion exchange chromatography are the best options. These affinity-purified antibodies, which lack unwanted anti-species cross-reactivity, are markedly superior to those obtained with standard purification methods. A blocking buffer that is formulated appropriately can also be used to increase assay sensitivity in addition to improving assay specificity. Active ingredients that are commonly used to control background noise include animal proteins, detergent, and salt. The levels of these ingredients have to be empirically evaluated as more is not always better and less may render them ineffective.
Quality of raw materials
Several guidelines exist on how to configure sandwich immunoassays (Boja et al. 2011, Ellington et al. 2010, Fuzery et al. 2013, xMAP Cookbook 2016) but it is not always obvious which guideline best applies to multiplex assays. In all cases, the quality of the raw materials, specifically the antibodies, and the intended use of the assay should be taken into consideration when determining the criteria of performance assessment. Inter-lot variability in raw materials can significantly impact the quality of an assay. This must be systematically managed to better control continuity in sample measurement. In developing sandwich immunoassays, the entire scaffold of antibody screening depends critically on the pairing of high-quality primary (capture) and secondary (detection) antibodies. Antibodies validated for single-plex immunoassays on one platform may not always display the same performance in a multiplex setting using a different technology. Monoclonal antibodies produced by hybridoma techniques are best used to confer the specificity of a sandwich assay. Either a monoclonal or a polyclonal antibody may be used as the detection antibody. A “mirror-sandwich” assay may be constructed using the same capture and detection antibody. However, this may only be robust for larger target antigens with multiple epitopes. As an alternative, recombinant antibodies could play a more critical role in the future as demand for high-sensitivity and -specificity antibodies increases. Other reagents in the buffer systems could also be critical to the overall quality and consistency of the end product, so establishing an orthogonal QC process is also critical.

Screening and qualification of antigens and antibodies
Unlike recombinant proteins, most commercial antibodies are not defined at the sequence level. Many of these antibodies are also not fully tested for cross-reactivity in a multiplex setting. In addition to brand awareness, it is critical to perform a thorough evaluation at the initial phase of raw material screening. These raw materials (antigens and antibodies) are typically subjected to four stages of evaluation when developing Bio-Plex Multiplex Immunoassays (Figure 1).
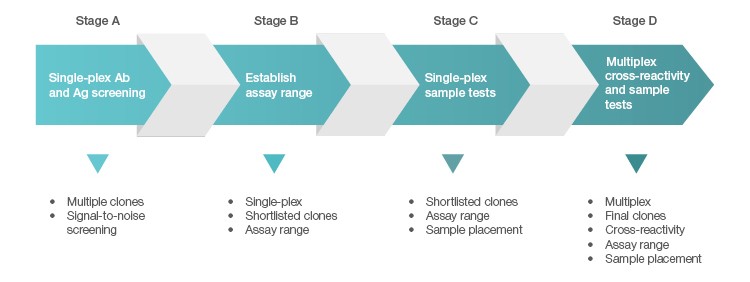
Fig. 1. Four-stage screening and qualification of antigens and antibodies.
Stage A is a rapid screening process carried out in single-plex format, with emphasis on identifying antibody pairs that produce the best signal-to-noise ratio in response to their respective antigens.
In Stage B, a short-listed antigen is expanded into a standard curve to assess a range in which robust assay recovery can be achieved. Recovery at the bottom of a curve is often dictated by the background of the assay. The lower the background, the more likely the curve can be extended out into the range of sub pg/ml.
Sample testing is included in Stage C to ensure optimal sample placement in addition to achieving a robust standard curve.
In Stage D, the selected targets are combined into a single multiplex blend to ensure these reagents can co-exist and retain their desired performance, including the ability to separate the healthy from the diseased samples with minimal interference from other targets within the multiplex panel. This is the most challenging part in designing multiplex assays. The odds of encountering assay cross-talk increase with the size of the panel developed.
Ultimately, the selection of antibodies for multiplex assays has to be stringent, and the buffer systems employed have to be robust enough to suppress cross-talk between assays.
Assay configuration
Sample placement is a key factor driving the configuration of a multiplex assay panel since an assay is useful only if it can measure samples with accuracy and precision. A systematic titration of the standard curve, along with its matching capture and detection antibody loadings, ensure that a working assay range has a robust capacity for sample measurement. An ideal standard curve extrapolates sample measurements from close to the middle of the curve where precision and accuracy are tight (Figure 2A). In cases where the sample readings are extrapolated close to the bottom of the curve (Figure 2B), a common strategy is to position the curve lower, without losing assay recovery. In cases where sample measurements are widely distributed along the curve (Figure 2C), we further expand the standard curve range along with optimization of sample dilution.
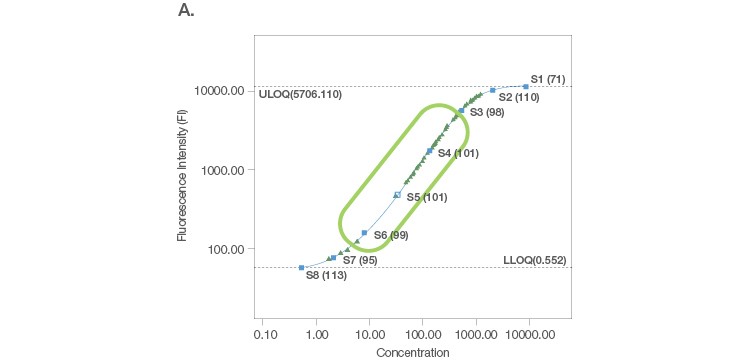
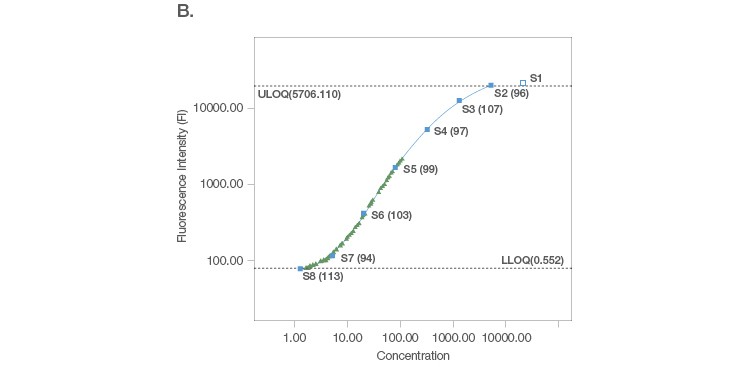
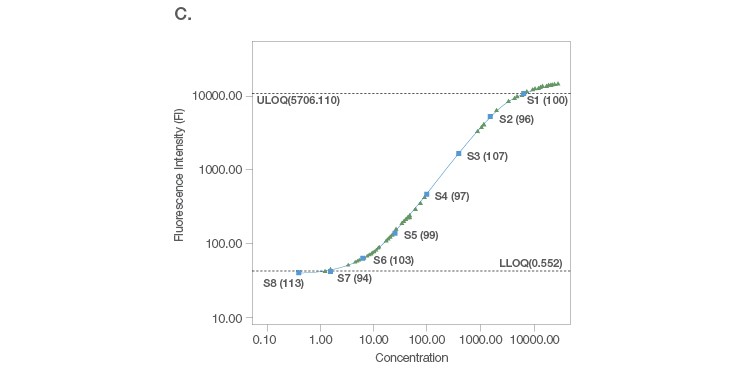
Fig. 2. Configuration of standard curves with sample placement using Bio-Plex Manager Software. A, an ideal standard curve where measurements are extrapolated from the middle of the curve. B, when sample measurements are extrapolated close to the bottom of the curve, the curve is positioned lower without losing assay recovery. C, when sample measurements are widely distributed along the curve, the standard curve range is expanded with sample dilution optimization.

There are many multiplex immunoassay manufacturers on the market and it is important for end users to ask the right questions to make an educated decision. Multiplex assay development and manufacturing is multifaceted and, in an ideal world, end users would only have to focus on their hypotheses and experiments and expect their results to be accurate, reproducible, and actionable. That is why, at Bio-Rad, we put actionable results at the center of our SMARTS approach: minimize end users’ effort, maximize their results. In order to maximize the benefit on performing multiplex immunoassays, end users must tailor the performance characteristics and capabilities of these assays to their specific studies. Key performance parameters to consider include sensitivity, multiplexiblity, actionable results, reproducibility, time, and specificity. Other factors that can impact the decision include the need to calibrate to a reference standard and to align assay results to an independent platform. Ultimately, the choice of a multiplex assay panel should be driven by its application, either for biomarker screening or validation. Proper validation of a commercial multiplex assay panel (to evaluate assay protocol, along with sample handling and preparation) is critical prior to any large-scale studies. Standardization of assay reagents and harmonization of the validation method will help to better control operator-to-operator and laboratory-to-laboratory variability. Within a given multiplex assay panel, it is reasonable to set performance specifications specific for each assay within the panel instead of using the blanket specifications provided by the assay vendor. These specifications can be further refined and tailored to the degree of precision or imprecision that an experimental study is able to tolerate. For users transitioning from an ELISA platform, it is critical to carry out cross-platform alignment studies specifically for the absolute measurement of cytokines. In many cases the quantitative discordance between ELISA and multiplex assays may be systematically harmonized by testing reference control samples in the alignment study. Standardization of assay reagents and harmonization of the validation method is the key to alignment between assay vendors and assay platforms.

Assay accuracy and matrix effects
Sample matrices commonly used for multiplex cytokine assays include serum and plasma. These sample types can consist of interfering/inhibitory components other than the cytokines of interest that are present near the LOD of the assays. These matrix components may hinder the quantitative results and affect the accuracy of sample readout. Linearity to dilution and parallelism are two very important assay performance parameters that are evaluated in every single assay. The linear response of a cytokine assay can be determined by replicate analysis of a series of spiked samples containing a known quantity of a recombinant cytokine in serum. An assay is considered to be accurate if the recovery of these spiked samples is between 70–130% (linearity dilution). This measures the degree of agreement of the test result to its theoretical true value. Parallelism is conducted with a positive serum sample that is serially diluted. The assay reagent is considered parallel to the sample matrix if the %CV of all the dilutions is less than 30% (Andreasson et al. 2015). The accuracy of the assay may be further evaluated and standardized using a reference standard, such as the NIBSC/WHO biological reference materials. This measures the degree of agreement of the test result to the reference value. In the absence of reference material, as with many non-human cytokine assays for which no NIBSC reference standards exist, comparison to an established reference assay (typically ELISA) may be the only option.
The accuracy of an assay can also be improved by minimizing matrix effects. This can be carried out by sample dilution using a buffer that is biologically comparable to the buffer that is used to construct the standard curve. A high sample dilution factor is permissible for cytokine markers that are present at high concentration or an assay that is highly sensitive. A high sample dilution will reduce the impact of matrix effect (improved linearity) in addition to reducing the requirement for sample volume. For cytokine assays that are less sensitive, along with the low abundance markers that are not readily detectable, the weight of improving sample detectability may fall on the buffer system. An ideal blocking buffer binds to all potential sites of nonspecific interaction, suppressing background, without altering or obscuring the epitope for antibody binding. Bovine and mouse serum have been found to be effective as blocking agents for heterophilic antibodies produced by the human immune system. The addition of these heterologous sera to the buffer system must be evaluated empirically via a matrix matching study to make sure they are biologically comparable to a given sample matrix.
Precision profile
A precision profile depicts the closeness of agreement between a series of sample measurements. The closer the result replicates, the more likely that the results will be repeatable in subsequent experiments. On Bio-Plex Manager Software, precision is derived from standard deviations (STDEV) and the coefficient of variation (CV) and reported in %CV. The %CV calculated using median fluorescence intensity (MFI) reflects between-well CV in the raw fluorescence values. These CV values are often tighter than the %CV reported using observed concentration (Obs Conc) and are best used to monitor between-well CV. Intra-Obs Conc %CV is dictated by the precision of the entire curve and is best used to judge the %CV in sample concentration measurements. Operator errors such as pipetting and using the incorrect shaking speed often contribute to high %CV values. Instrument error can be prevented by implementing periodic preventative maintenance. Automation in liquid handling in these assays is a major driver for an improvement in assay precision.
Quality control
To maintain the quality of developing these assays, the utility of quality controls should be emphasized, both at the production level, and for the end users. The scope of a quality control performed during the production phase should be in alignment with the reagent control manufactured for the end users. This ensures seamless evaluation of assay performance in the hands of the end users. In addition to the reagent controls provided in a commercial kit, a set of user-defined reference samples (prepared using a specific matrix comparable to the test specimen) may also be used to monitor operator and/or inter-plate variability.

Product design and performance
In addition to significant cost saving, the use of these multiplex assays also shortens time to results. The ability to concurrently generate a large set of data from a single plate run (78-well x 48 assays = 3,744 data points) confers tremendous time savings. The design of a simplified assay workflow with the use of ready-to-use reagents and shorter assay incubation and data acquisition time, will further reduce time to results. A favorable time to results will improve workload capacity, permitting more data to be generated in a single day. To this end, automation also plays a key role in faster time to results. Automation may not always be economical (especially for laboratories with lower capacity), but it is critically essential to achieve higher throughput and faster time to results and improve assay performance. Automation in sample preparation, liquid handling (dispensing, aspirating, and washing), and incubation will increase productivity in laboratories.
Product design
A multiplex panel must generate results that are accurate and reproducible and can lead end users to greater insights about their studies. The key element in the development of multiplex immunoassays is selection of biologically relevant biomarkers. Integration of data science or bioinformatics with expert knowledge-driven approaches to develop biologically relevant assays enables end users to assess the complexity of multifactorial diseases such as cancers, stroke, and diabetes mellitus with greater precision. Improvement in translational research is fueled by biomarker discovery, drug development, and the emerging demands for multiplex point-of-care testing. The advent of multiplex technologies has sped up the identification of biomarkers for many diseases. Composite profiling of biomarkers makes good financial as well as scientific sense from any perspective.

Controlling assay specificity by suppressing assay interference
Interference in immunoassays is a persistent and perhaps underestimated issue despite attempts by assay manufacturers to address it. Assay interference can be easily studied and resolved in a single-plex sandwich assay. This effect, however, is magnified in a multiplex setting due to the combinatorial interaction of each antibody with other ligands. This is the primary source of assay cross-talk in multiplex immunoassays. Assay cross-talk can be reagent driven or sample driven or a combination of both. Reagent-driven cross-talk is a critical factor in multiplex bead array sandwich assays. It was mathematically modeled by Juncker et al. (2014) and Pla-Roca (2012), who introduced the concept of liability pairs in his analysis of vulnerability to cross-reactivity in multiplex sandwich assays. Liability pairs designate the combination of antibody-antibody, antibody-antigen, or antigen-antigen in single cross-reactive binding among proteins that translate into a false positive signal or background noise. These nonspecific signals are systematically investigated via single detection and single antigen cross-reactivity studies. Single detection cross-reactivity studies are performed in the presence of multiplex antigens and capture antibodies, where the single detection antibody is evaluated for cross-reactivity (Figure 3A). Single antigen cross-reactivity studies are performed in the presence of multiplex capture and detection antibodies, where the single antigen is evaluated (Figure 3B).
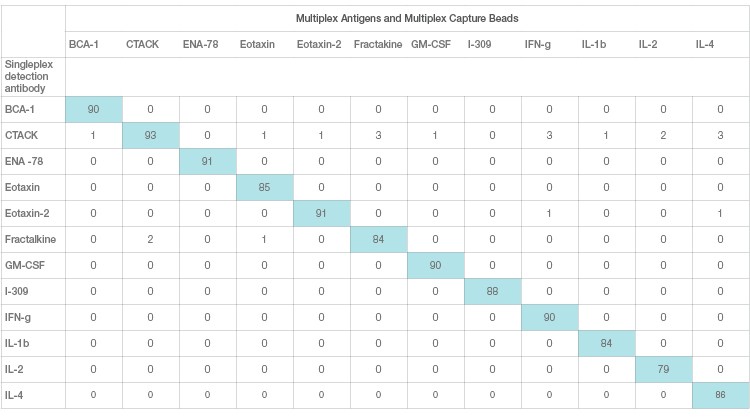
Fig. 3A. Single detection cross-reactivity. Each row represents one cross-reactivity experiment where a single detection antibody is assayed with multiplex Ag and beads. A specific detection should only recognize the matching pair and not any other capture antibodies.
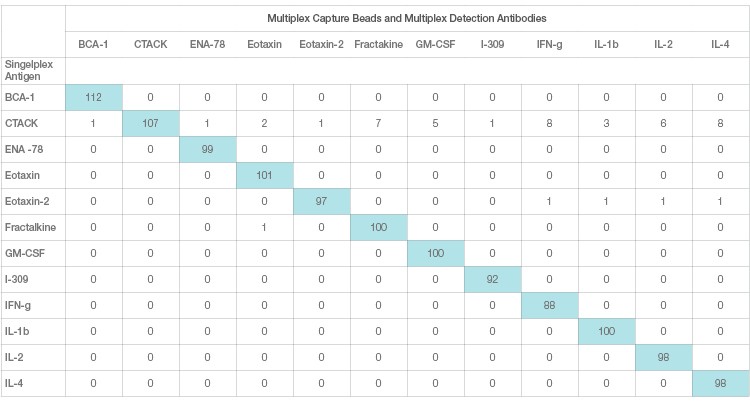
Fig. 3B. Single antigen cross-reactivity. Each row represents one cross-reactivity experiment where only one antigen is assayed with multiplex beads and detection. A specific recombinant antigen should only be recognized by the matching antibody pair and not any other antibodies.
Sample-driven cross-reactivity can have an additive effect on reagent-driven cross-reactivity. Endogenous interfering substances, including interfering antibodies, can be present in both healthy and diseased human plasma samples. Exogenous interfering substances include sample collection additives, carrier proteins, drugs, hemolysis, and lipemia (Schiettecatte et al. 2012). Removal or reduction of these interfering components is commonly carried out by specific sample clean up and/or the addition of specific blocking agents into the sample treatment buffer. The design of a universal blocking buffer for multiplex assays is an ambitious task. Often, a single blocking agent is not sufficient to suppress interference within a multiplex setting. Addition in the reaction mixture of low concentrations of serum or immunoglobulin from the same species as the antibody reagents can neutralize or inhibit interference in some samples. Non-immune serum, polyclonal IgG, polymerized IgG, non-immune mouse monoclonal, or fragments of IgG (Fc, Fab, F(ab’)2) from the same species used to raise the reagent antibodies are commonly used as blocking agents (Schiettecatte et al. 2012). Ultimately, a dedicated titration scheme for all the antibodies will be required to overcome cross-talk between assays.
Current status and future direction
Currently many commercially available multiplex cytokine immunoassays vary in their ability to measure serum and/or plasma cytokines (Alex et al. 2009, Berthoud et al. 2011, Breen et al. 2011, Christiansson et al. 2014). Often, the evaluation and comparison between different commercial assays are limited by the lack of a reference standard. Despite these known hurdles, the measurement of multiple cytokines simultaneously within a specimen still enables the assessment of relative levels of cytokines within a system. Multiplex assays have become widely used in basic research because of their many advantages over single-plex ELISA assays. The benefits include high throughput, lower cost, and low consumption of sample volume. These assays are reaching a stage of maturity where, with proper validation, they can be successfully implemented in clinical development programs. While the utility of multiplex cytokine assays as diagnostic and prognostic tools in diverse autoimmune, inflammatory, infectious, and malignant conditions is still limited, its potential is expanding. Relative to conventional ELISA assays, many of these assays offer a wider assay range and have become increasingly more sensitive (down to the single digit picogram/ml range). Aside from the Luminex xMAP platform, the most desired immunoassay module researchers want access to is an open platform with enhanced speed and an automated solution for generic immunoassay applications that can be adapted for future requirement changes. At present, suspension immunoassay is the prevailing technology for FDA-cleared multiplex protein measurements (Tighe et al. 2015).
Learn more about Bio-Plex Multiplex Immunoassay technology.
References
Andreasson U et al. (2015). A practical guide to immunoassay method validation. Front Neurol 6, 179.
Alex P et al. (2009). Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis 15, 341–352.
Berthoud TK et al. (2011). Comparison of commercial kits to measure cytokine responses to Plasmodium falciparum by multiplex microsphere suspension array technology. Malar J 10, 115.
Boja ES et al. (2011). The journey to regulation of protein-based multiplex quantitative assays. Clin Chem 57, 560–567.
Breen EC et al. (2011). Multisite comparison of high-sensitivity multiplex cytokine assays. Clin Vaccine Immunol 18, 1,229–1,242.
Christiansson L et al. (2014). The use of multiplex platforms for absolute and relative protein quantification of clinical material. EuPA Open Proteom 3, 37–47.
Comley J (2014). Immunoassay automation: hands-free platforms set to change the workflow in research labs. ddw-online.com/screening/p274234-immunoassay-automation:-hands-free-platforms-set-to-change-the-workflow-in-research-labs.html, accessed June 18, 2018.
Ellington AA et al. (2010). Antibody-based protein multiplex platforms: technical and operational challenges. Clin Chem 56, 186–193.
Etzioni R et al. (2003) Combining biomarkers to detect disease with application to prostate cancer. Biostatistics 4, 523–538.
Fuzery AK et al. (2013). Translation of proteomic biomarkers into FDA approved cancer diagnostics: issues and challenges. Clin Proteomics 10, 13.
Hsu MJ et al. (2014). Biomarker selection for medical diagnosis using the partial area under the ROC curve. BMC Res Notes 7, 25.
Juncker D et al. (2014) Cross-reactivity in antibody microarrays and multiplexed sandwich assays: shedding light on the dark side of multiplexing. Curr Opin Chem Biol 18, 29–37.
Pla-Roca M et al. (2012). Antibody colocalization microarray: a scalable technology for multiplex protein analysis in complex samples. Mol Cell Proteomics 11, M111011460.
Schiettecatte J et al. (2012). Interferences in immunoassays. intechopen.com/books/advances-in-immunoassay-technology/interference-in-immunoassays, accessed June 18, 2018.
Tighe PJ et al. (2015). ELISA in the multiplex era: Potentials and pitfalls. Proteomics Clin Appl 9, 406–422.
Turner MD et al. (2014). Cytokines and chemokines: At the crossroads of cell signaling and inflammatory disease. Biochim Biophys Acta 1843, 2,563–2,582.
Williams FMK (2009). Biomarkers: in combination they may do better. Arthritis Res Ther 11, 130.
xMAP Cookbook (2016). info.luminexcorp.com/en-us/research/download-the-xmap-cookbook, accessed June 18, 2018.
Bio-Rad and Bio-Plex are trademarks of Bio-Rad Laboratories, Inc. in certain jurisdictions. All trademarks used herein are the property of their respective owner.