The power to edit a gene is the power to change its function and with it the biology of a cell — or even a human being. This realization has driven countless scientists and laboratories to develop ever improving genome editing tools; some with the goal of better understanding biology, others with the goal of improving crops or developing more effective therapeutics.
The earliest forms of gene editing were based on homologous recombination, the ability to exchange nucleotide sequences between similar pieces of DNA. This worked only in a small subset of model systems, such as mouse and yeast, where the frequency of recombination was high enough to make it feasible. Even in those systems, this process was very laborious, requiring stringent and time-consuming selection protocols. The insight that propelled the field of genome editing forward was that the frequency of homologous recombination could be greatly increased by using an endonuclease to generate a double stranded DNA break (DSB). Over the past 20 years this discovery has led to the development of a range of genome editing technologies that have expedited basic research and made their way into clinical trials (Table 1).
Table 1. Comparison of gene editing techniques.
| Meganucleases | Zinc Finger Nucleases | TALENs | CRISPR-Cas9 | |
| Target site length | 14–40 bp | 18–36 bp per ZFN pair | 28–40 bp per TALEN pair | 19–22 bp |
| Off-target effects | Some mismatches are tolerated | Some mismatches are tolerated | Some mismatches are tolerated | Tolerant even of several consecutive mismatches |
| Targeting constraints | Reengineering for new specificities is challenging | G-rich regions are challenging | 5ˈ T for each TALEN is required | PAM is required |
| Ease of design | Extremely difficult | Difficult | Moderate | Easy |
| Multiplexing | Challenging | Challenging | Challenging | Easy |
| Ease of in vivo delivery | Small size allows use of many viral vectors | Small size allows use of many viral vectors | Large size of each TALEN limits viral vectors, and repetitive sequences lead to unwanted recombinations | S. pyogenes Cas9 is too large for smaller capacity viral vectors |
| Ease of ex vivo delivery | Relatively easy | Relatively easy | Relatively easy | Relatively easy |
Meganucleases and megaTALs
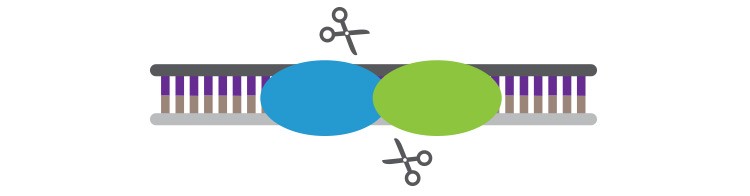
Meganucleases are highly specific and easy to deliver to cells but difficult to redesign for new targets.
Meganucleases, also called homing endonucleases, were the first targeted endonucleases discovered and appeared promising for gene editing because their 14–40 bp recognition sites are rarely found more than once in a genome. Their small size is compatible with most cell delivery strategies, and only a single protein is required for highly efficient gene editing. What has kept meganucleases from becoming widely used is that they are difficult to engineer for new specificities.
Creating new meganucleases requires advanced protein engineering, such as altering specificities of naturally occurring meganucleases through a series of difficult-to-predict mutations or creating chimeric proteins that combine the active sites of two naturally occurring meganucleases. Cellectis (Paris, France) has developed an extensive library of protein domains that can be combined to form meganucleases with a vast number of different specificities while Precision BioSciences’ (Durham, NC) proprietary ARCUS technology utilizes a fully synthetic enzyme similar to meganucleases that can be more easily reengineered to accommodate new target specificities. The requirement for such advanced protein engineering, however, has made meganucleases less accessible and broadly used than TALENs or CRISPR-Cas9 (discussed below).
Related to meganucleases and TALENs, megaTALs combine the easy-to-design DNA binding domains from TALENs with the high cleavage efficiency of meganucleases in a single chimeric protein. Both megaTALs and meganucleases are very promising for therapeutic applications because of their compact size and high cleavage efficiency but are less frequently used for research applications due to design challenges.
Zinc Finger Nucleases (ZNFs)
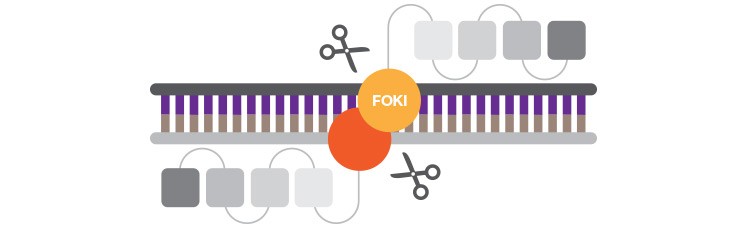
Although first to clinical trials, zinc finger nucleases have been more difficult to design than anticipated given their modular nature.
Zinc finger nucleases (ZFNs) were the first targetable endonucleases that were relatively easy to program. These artificial nucleases combine DNA binding domains called zinc fingers (ZFs), normally used to target transcription factors to the genes they regulate, with a non-specific endonuclease, FokI. Each ZF specifically recognizes three to four bases of DNA. By linking multiple ZFs of known specificity, FokI nuclease can be targeted to a sequence of interest. To generate DSBs a pair of ZFNs needs to be designed to flank the desired cut site. At first promising because of their seemingly modular design, ZFNs have proven to be more difficult to design than anticipated because individual ZFs are not truly independent of one another — base pair recognition of one ZF is affected by neighboring ZFs in a difficult-to-predict manner. Highly specific ZFNs, however, can be successfully created using context-dependent design methods such as oligomerized pool engineering (OPEN) and context-dependent assembly (CoDA). Sangamo BioSciences (Richmond, CA) has successfully designed ZFNs, for HIV/AIDS, hemophilia B, Hurler syndrome, and Hunter syndrome gene therapy, that are currently in phase 1 and 2 clinical trials.
Transcription activator-like effector nucleases (TALENs)
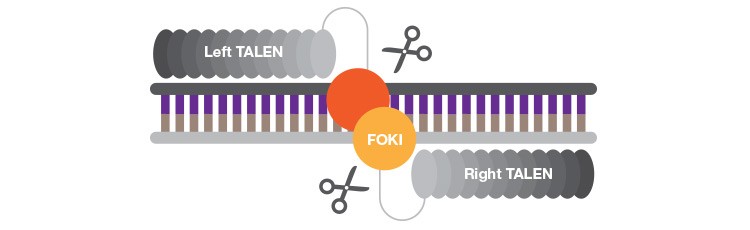
TALENs are truly modular and not limited by sequence requirements, but their large size makes cell delivery challenging.
TALENs circumvent the design difficulties that have slowed the wide adoption of ZFNs. Like ZFNs, TALENs utilize the non-specific nuclease FokI and act in pairs. Unlike ZFNs, TALENs are truly modular. TALEN DNA binding domains consist of repeating units of 33–35 amino acids, each of which contains two amino acids at defined positions that confer specificity for a single nucleotide. A one-to-one relationship exists between these guiding amino acid pairs and the four DNA bases, making design of new TALENs far simpler than design of ZFNs. A crucial advantage of TALENs over all other programmable nucleases is that they can be designed to bind virtually any target sequence; the only requirement is a thymidine at the 5′ end of the targeted sequence.
However, because each 33–35 amino acid repeat recognizes only a single base pair, TALENs are significantly larger than ZFNs or meganucleases. Most TALENs consist of up to 20 repeating modules, which limits the vectors available to deliver them into target cells. The large stretches of repetitive sequences that characterize TALENs can also lead to undesired recombination events that can inactivate or redirect a TALEN and pose a significant challenge in assembly for traditional molecular biology techniques. Several methods have been developed to simplify assembly of TALENs, including the popular one-step Golden Gate system that has been used to build a TALEN library that targets 18,740 human protein coding genes (Kim et al. 2013). TALENs have shown equal promise in clinical applications; after successfully reversing one patient’s cancer, a second child has been treated at Great Ormond Street Hospital for Children in London using Cellectis’s CAR T-cell therapy for pediatric acute B lymphoblastic leukemia (Hirschler 2016). CRISPR startup Editas Medicine (Cambridge, MA) is also continuing to leverage TALEN technology in addition to their more publicized CRISPR development.
Clustered regularly interspaced palindromic repeats (CRISPR)-Cas9 System
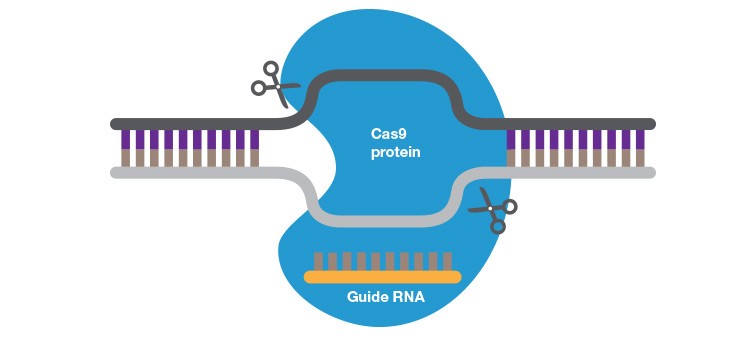
Easy to design and amenable to multiplexing, CRISPR-Cas9 is revolutionizing gene editing, but specificity concerns remain to be resolved.
CRISPR-Cas9 is the simplest gene editing technique to date and has truly revolutionized the field of gene editing. Based on the adaptive immune system of bacteria and archaea, the CRISPR-Cas9 system uses short RNAs to direct Cas9 nuclease to target DNA sequences. Cas9, like the FokI endonuclease utilized by ZFNs and TALENs, is a non-specific endonuclease that creates DSBs. What has set CRISPR apart from other gene editing techniques is its simplicity. Because it uses RNA rather than a protein to target nuclease activity, CRISPR-Cas9 can be retargeted by simply synthesizing or ordering a new guide RNA (gRNA). The RNA guide also makes this technique amenable to high order multiplexing; large-scale screens using hundreds or even thousands of gRNAs in a single experiment have been performed. Complex animal models that once took years to generate are now completed in just a few months.
This technique has democratized gene editing, allowing small research labs as well as large government or pharmaceutical labs to edit genes and to clear key regulatory hurdles in preparation for first clinical trials. Chinese scientists, for example, received approval in July to commence clinical trials using CRISPR-engineered CAR T-cells to treat patients with metastatic non–small cell lung cancer at Sichuan University’s West China Hospital in Chengdu. The University of Pennsylvania has been approved by a federal biosafety and ethics panel to conduct the first CRISPR clinical trial in the United States. This study aims to determine whether CRISPR-engineered CAR T-cells are safe for use in the clinic. The University is now awaiting approval by the U.S. Food and Drug Administration and the medical centers that will carry out the trial. Editas Medicine is promising clinical trials for a rare form of blindness in 2017.
Maximizing the utility of CRISPR-Cas9
The current CRISPR-Cas9 system, like the other gene editing technologies discussed here, still poses a number of formidable challenges, including its size. The commonly used Streptococcus pyogenes Cas9, for example, is too large to be accommodated by adeno-associated virus (AAV) delivery vectors, which are popular for gene therapy because they integrate into a known “safe harbor” site of the genome and have an excellent safety profile. The recent discovery of a Cas9 variant from S. aureus that is 1,000 kb shorter than the S. pyogenes gene has addressed this limitation (Ran et al. 2015) and has improved potential for efficient therapeutic delivery of the CRISPR-Cas9 system into target cells.
A second major constraint of the CRISPR-Cas9 system is its requirement for a species-specific sequence, called a protospacer adjacent motif (PAM), near the edit site. This limits the sequences that can be edited using a single organism’s Cas9. Significant efforts have been put into expanding the sequences that can be edited using CRISPR-Cas9 by characterizing Cas9 from a variety of organisms. These studies led to the discovery of a new protein, Cpf1, that not only has different sequence requirements but also creates DSBs with overhangs instead of the blunt ends created by Cas9. These overhangs can be used to create a more controlled insertion of DNA during gene editing (Kim et al. 2016, Kleinstiver et al. 2016).
Lastly, off-target edits are a concern for CRISPR-Cas9 because it tolerates positional as well as multiple consecutive mismatches. Significant progress has been made in engineering Cas9 and gRNA variants that increase CRISPR’s specificity, including engineering of high-fidelity Cas9 variants (Kleinstiver et al. 2015, Slaymaker 2015), Cas9 nickase mutants (Ran et al. 2013), and synthetic gRNAs (Kelley 2016).
Gene editing beyond CRISPR
An entirely new gene editing system was recently presented in Nature Biotechnology. Although the results still need to be reproduced, this new system, based on Argonaute proteins — best known for their involvement in RNA interference — uses a protein called NgAgo to generate DSBs. Rather than using a gRNA it uses DNA to guide nuclease activity, which eliminates the need to transcribe gRNAs in vitro or express them from a plasmid. This system also promises to be more flexible than CRISPR-Cas9 because it is not limited by PAMs and is more accurate due to its low tolerance for mismatches between guide DNA and target sequence (Gao et al. 2016).
Another gene editing technology, based on the CRISPR-Cas9 system, provides the ability to edit without cutting. When Cas9 creates DSBs it often creates random insertions and deletions (indels) through nonhomologous end-joining (NHEJ). Those who want to introduce new sequences or do more than simply disable a gene have to rely on the much less frequent homology directed repair (HDR) pathway. By generating a non-cutting version of Cas9 and fusing it to an enzyme that converts cytidines to uredines, C→T point mutations could be generated without risking generation of NHEJ-mediated indels. Hinting at the therapeutic potential of this technology, mutations associated with Alzheimer’s disease could be corrected in cultured mouse cells with efficiencies up to 75% (Komor et al. 2016). A similar approach was published separately in Science (Nishida et al. 2016).
Findings like these, and work in laboratories such as those of CRISPR pioneers George Church and Feng Zhang, who are hoping to add enzymes like integrases and recombinases to our repertoire of genome editing tools, suggest that we may have only just begun to explore the potential of genome editing.
References
Gao F et al. (2016). DNA-guided genome editing using the Natronobacterium gregoryu Argonaute. Nature Biotechnol 34, 768–773.
Hirschler B (2016). Second baby gets Cellectis “designer” cells to clear leukemia. Reuters, May 6. reuters.com/article/us-health-celltherapy-idUSKCN0XX1F7. Accessed Sept. 15, 2016.
Kelley ML et al. (2016). Versatility of chemically synthesized guide RNAs for CRISPR-Cas9 genome editing. J Biotechnol 233, 74–83.
Kim Y et al. (2013). A library of TAL effector nucleases spanning the human genome. Nat Biotechnol 3, 251–258.
Kim D et al. (2016). Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol 8, 863–868.
Kleinstiver BP et al. (2015). High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495.
Kleinstiver BP et al. (2016). Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol 8, 869–874.
Komor AC et al. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424.
Nishida K et al. (2016). Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science pii: aaf8729 [Epub ahead of print].
Ran FA et al. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1390.
Ran FA et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191.
Slaymaker IM et al. (2015). Rational engineered Cas9 nucleases with improved specificity. Science 351, 84–88.


