Abstract
Genome editing has become a widely used tool and allows specific modification of almost any cell’s genome. However, the often low (<5%) frequency of gene edits, in particular for some primary or induced pluripotent stem cell types, hinders facile and reliable detection by gel- or sequencing-based methods, or requires laborious clonal isolation of edited cells. Here we introduce an ultra-sensitive, cost-efficient Droplet Digital PCR–based method for the detection of HDR and NHEJ alleles generated using the CRISPR/Cas9 and TALEN genome editing systems.
Figures
|
Figure 1. Droplet group definitions for drop-off assay analysis. |
Figure 2. Assay design for detection of small (<20 bp) and large (>20 bp) HDR edits. |
|
Figure 3. A three-step cycling protocol improves amplification of long amplicons. |
Figure 4. Assay design for the detection of NHEJ edits. |
|
Figure 5. Quantification of HDR alleles in synthetic templates and edited HEK293T cells. |
Figure 6. Quantification of NEHJ alleles in synthetic templates and edited HEK293T cells. |
|
Figure 7. Quantitative comparison of gRNA usage |
|






Introduction
Genome editing tools including TALENs and the CRISPR/Cas9 system have revolutionized our ability to edit the genome of any cell, including human induced pluripotent stem cells (iPSCs). Sequence-specific nucleases induce double-strand breaks or nicks at target sites, activating the DNA repair pathways of non-homologous end joining (NHEJ) and homology-directed repair (HDR). NHEJ produces small insertions or deletions (indels) and is useful for disrupting gene function. HDR can induce precise gene repair of one to thousands of base pairs in the presence of a homologous donor molecule, allowing for correction of point mutations and introduction of exogenous sequences. Genome editing is an increasingly common part of the molecular biologist’s toolkit and is being actively developed for therapeutic indications.
While powerful, the efficiency of gene editing is cell type – dependent and often low (<5%), particularly in primary cells or iPSCs. Sanger sequencing and gel-based methods lack the sensitivity required to detect such events in non-clonal cell populations. Detection by next-generation sequencing (NGS) presents hurdles in terms of cost and workflow, and NGS sample prep can introduce bias for or against edited alleles.
Droplet Digital PCR (ddPCR™) enables ultra-sensitive absolute quantification of genome editing events. Here we introduce ddPCR assay strategies for the detection of HDR- and NHEJedited alleles. Using these assays we can detect alleles in edited samples present at frequencies of less than 0.5%. These methods are useful for ultra-sensitive detection of edited alleles and offer a rapid, low-cost readout for technical optimization of genome editing protocols.
Methods
Genome Editing
The disease-relevant loci RBM20 and GRN were targeted for editing in HEK293T cells using TALEN, wild type (WT)-Cas9, dCas9-FokI, or Cas9-nickase strategies. TALENs were constructed using the Voytas laboratory’s Golden Gate assembly system (Cermak et al. 2011) and the MR015 backbone vector (gift from M. Porteus and M. Rahdar, Stanford University). Guide RNAs (gRNAs) were cloned into the Zhang laboratory’s pX335 (nickase) and pX330 (nuclease) vectors (Cong et al. 2013 and Ran et al. 2013).
Droplet Digital PCR
The QX200™ Droplet Digital PCR System (Bio-Rad Laboratories) was used as instructed by the manufacturer. ddPCR reactions were assembled using standard protocols: ddPCR Supermix for Probes (no dUTP) (Bio-Rad Laboratories) was combined with 5–125 ng of sample, 1 μl of 20x assay mix (1x primers at 900 nM each, 1x probes at 250 nM each), 4 units of restriction enzyme (HaeIII for RBM20 or MseI for GRN), and water for a final reaction volume of 20 μl. Reactions were converted into approximately 20,000 one-nanoliter droplets using the QX200 Droplet Generator and transferred to a 96-well plate for thermal cycling per manufacturer recommendation for this supermix. For GRN assays, a discrete 2 minute extension step at 72°C was included, generating a three-step thermal cycling protocol that accommodates a longer amplicon (252 bp). After thermal cycling, droplets were read on the QX200 Droplet Reader and assigned as positive or negative based on fluorescence amplitude.
Allele Quantification
For HDR assays, cluster thresholding and quantification was performed using QuantaSoft™ Software. For NHEJ drop-off assay analysis, cluster thresholds were assigned using QuantaSoft and a modified data analysis strategy was used for quantification.
Drop-off assays result in a two-dimensional plot with three clusters: droplets with no template (Nempty), single positive droplets containing NHEJ alleles (NNHEJ), and droplets that contain WT Alleles (NWT), which are positive for both FAM and HEX signal and appear on the 45° axis. (Figure 1). At low sample loading, for example ~2,000 copies/well (~5 ng human genomic DNA), most NHEJ mutant copies will occur in single positive (FAM-only) droplets. However, as sample loading or NHEJ fractional abundance increases, double positive WT+NHEJ droplets appear as a “tail” extending leftward from the WT-only cluster. In most cases, separation between WT-only and double positive droplet clusters is inadequate for robust thresholding because a distinct boundary is absent. For this reason, and because the mathematical analysis described below accounts for it, the WT+NHEJ double positive tail is grouped with the WT-only (green) cluster as part of the WT+ droplet group. It is not advisable to eyeball a threshold for double positives; this is subjective and will likely be incorrect. Future iterations of QuantaSoft Software will automate this analysis.
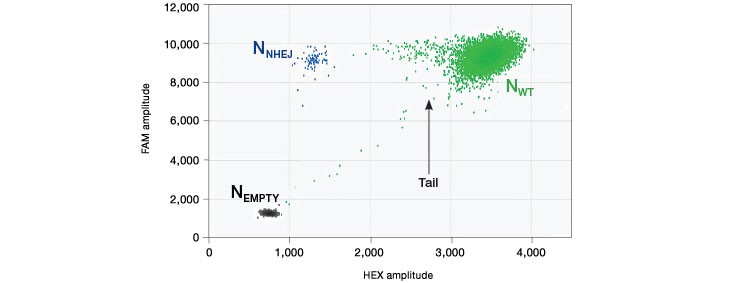
Fig. 1. Droplet group definitions for drop-off assay analysis.
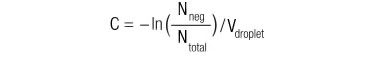
| Nneg | = the number of droplets that do not contain species of interest |
| Ntotal | = the total number of droplets |
| C | = concentration |
| Vdroplet | = droplet volume |
Because WT and WT+NHEJ droplet populations cannot be easily separated, it becomes difficult to define Nneg and Ntotal. To quantify NHEJ and WT copies from a drop-off assay, an
appropriate subset of droplets is used to calculate Nneg and Ntotal where:
- Nempty = the number of droplets in the double negative cluster labeled “empty” (black)
- NNHEJ = the number of droplets in the cluster labeled “NHEJ” (blue)
- NWT = the total number of droplets in the cluster labeled “WT+” (green)
For NHEJ quantification:
| Nneg | = Nempty |
| Ntotal | = Nempty + NNHEJ |
For WT quantification:
| Nneg | = Nempty + NNHEJ |
| Ntotal | = Nempty + NNHEJ + NWT |
For NHEJ quantification, note that only NHEJ single positive droplets are used for analysis. New analysis tools are in development that will allow NHEJ quantification directly from properly thresholded QLP files.
Results
HDR Edit Detection Using Droplet Digital PCR
We developed assay strategies for the sensitive detection of small (point mutations and <20 bp) and larger (>20 bp) precise edits introduced via HDR (Figure 2). Using an allelic discrimination TaqMan Assay and Droplet Digital PCR, Miyaoka et al. (2014) previously demonstrated ultra-sensitive detection of edit-induced point mutations in iPSC cells, with detection sensitivity as low as 1 in 5,000 (0.02%). This approach uses one primer pair that amplifies the intended edit region and two competitive probes labeled with FAM and HEX that are designed to bind edited (mutant) or WT sequences, respectively. Note that one primer is positioned outside the donor molecule sequence to ensure quantification of integrated edited sequences (Figure 2A).
Larger exogenous sequences, for example GFP or FLAG tags, can also be introduced by HDR-mediated editing. To detect these rare integrated alleles, one reverse primer and probe (FAM) is designed near the 5′ end of the intended fusion site inside the exogenous sequence (Figure 2B). The forward primer is gene-specific and sits upstream of the intended fusion site but outside of the homology arm sequence to ensure quantification of integrated alleles only. To count the total number of genomes interrogated, a standard reference assay, such as the PrimePCR™ ddPCR Copy Number Assay for RPP30 in HEX (assay ID dHsaCP1000485), may be used in duplex if amplicon lengths are similar or in a second well if lengths differ. Using a reference assay of amplicon length similar to the designed edit detection target assay avoids biased amplification in double positive droplets and serves to control for sample integrity.
A.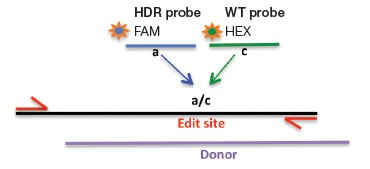
B.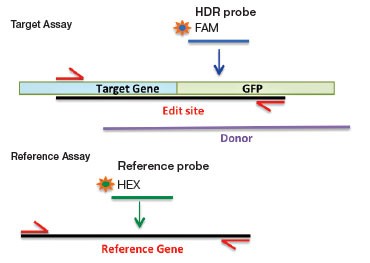
Fig. 2. A: HDR Edit Detection Assay (Point and Small (<20 bp) Mutations). B: HDR Edit Detection Assay (Large (>20 bp) Insertions)
Some donor molecules have long homology arms, which influences edit detection assay amplicon length. We found that amplification efficiency is reduced when amplicon lengths exceed 200 bp. This manifests as reduced amplitude of positive droplets. A three-step thermal cycling protocol with increased extension time (Table 1) improved performance significantly in those cases (Figure 3).
A.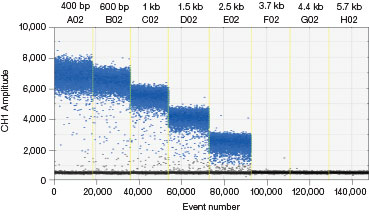
B.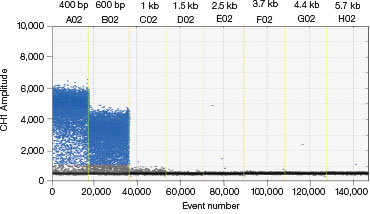
Fig. 3. A three-step thermal cycling protocol with long extension times improves amplification of long amplicons. A plasmid was quantified keeping one primer constant but varying the amplicon length by changing the location of the forward primer. Longer amplicons amplified more efficiently with a three-step protocol with 2 minute extension (A) compared to a standard two-step protocol (B), as evidenced by a significant improvement in separation between negative (blue) and positive (grey) droplet populations.
Table 1. Recommended three-step extension times for long amplicons. Length windows are approximate, will vary with sequence, and should be validated empirically. Use of ddPCR Supermix for Probes (no dUTP) and the deep-well C1000™ Thermal Cyler is advised.
| Amplicon Length, kb | Extension Temp,°C | Extension Time, min |
| 150–1.5 | 72 | 2 |
| 1.5–2.5 | 72 | 4 |
| 2.5–4 | 72 | 6 |
A.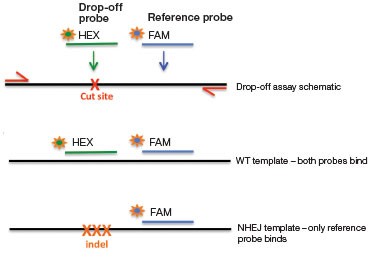
B.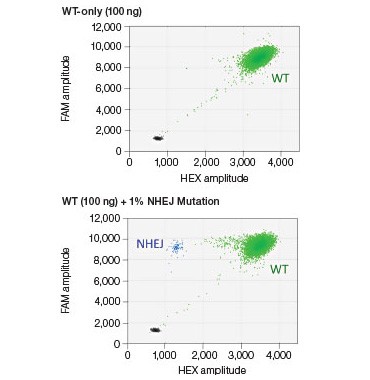
Fig. 4. NHEJ drop-off assay. A, primer and probe design for the NHEJ drop-off assay; B, 2-D droplet fluorescence intesity plots of an NHEJ drop-off assay using WT-only DNA and a Gene Fragment NHEJ mutation-positive control (WT + 1% gBlocks Gene Fragment containing a one base pair deletion at the predicted cut site). The WT cluster (green) is positive for both FAM and HEX, while the NHEJ mutant single-positive droplets (blue) are positive for FAM only.
NHEJ Edit Detection Using Droplet Digital PCR
The introduction of indels can be a desired outcome of NHEJ when gene inactivation (knockout) is intended. NHEJ-induced indel alleles can also occur when specific edits mediated by HDR using a donor template are sought. In either case, NHEJ indel mutations can provide a readout of genome editing nuclease activity during genome editing.
The challenge with detecting NHEJ edits is that a number of different alleles can be generated from a single treatment of cells with genome editing nucleases (Ran et al. 2013). One edited sample can thus contain NHEJ alleles with deletions and/or insertions of varying sizes. To quantify the indel alleles generated, we used a drop-off assay strategy. This is an assay
that identifies mutated alleles based on the absence of a WT signal.
In this assay, one primer pair amplifies the sequence that includes the predicted cut site (Figure 4A). The cut site could be, for instance, a site 3 bp upstream of a CRISPR protospacer-adjacent motif (5′-NGG PAM) or the sequence between two TALEN sites. A FAM-labeled reference probe distant from the site of potential NHEJ is designed to count all copies of the amplicon. A HEX-labeled drop-off probe is designed to bind WT DNA and targets the predicted cut site. This probe will not bind, or will “drop off,” when the allele has an insertion or deletion at this site. It is the loss of this HEX signal (while maintaining FAM signal) that indicates presence of an NHEJ allele (Figure 4B). Since this assay produces three-cluster 2-D fluorescence intensity plots, the resulting data cannot be directly quantified by QuantaSoft Software, up to and including versions 1.7 and 1.8. Instructions for quantification are presented in the Methods section. An automated analysis mode for this type of experiment will be included in the next version of the software.
High-Sensitivity Quantification of HDR Alleles
Dilated cardiomyopathy and sudden early death is associated with heterozygous germline mutations in RBM20, which encodes an RNA-binding motif protein (OMIM: 613172). Sensitive detection of the RBM20 R636S C>A pathogenic point mutation was performed using an allelic discrimination edit detection assay. RBM20 R636S was detected down to frequencies of less than 0.05% in 100 ng WT DNA (Promega Corporation) spiked with a 2x serial dilution of gBlocks Fragment synthetic template containing the C>A substitution (Figure 5A). The same BM20 R636S edit detection assay was used on HEK293T cells that had been treated with either CRISPR/Cas9- or TALEN-based editing reagents to introduce the C>A mutation for cell-based studies. Cell pools with <1% edited RBM20 alleles were easily identified (Figure 5B).
A.
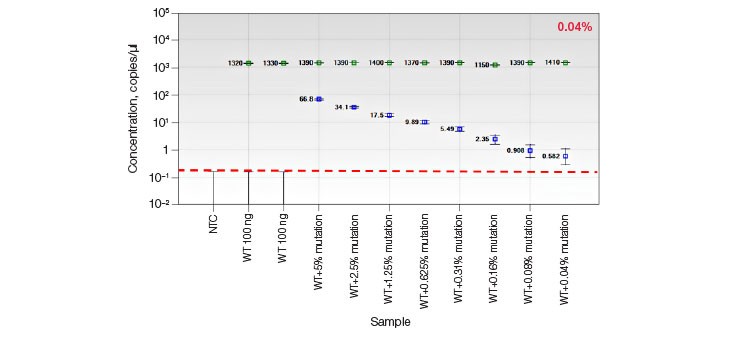
B.
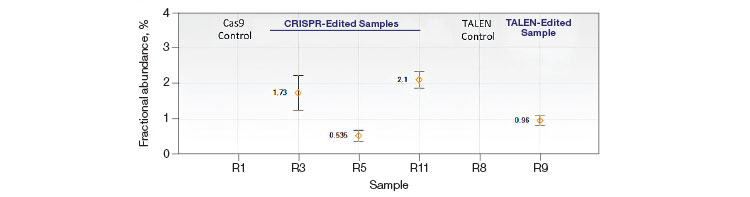
Fig. 5. Quantification of HDR alleles in synthetic mutant templates and edited HEK293T cells. A, single-well measurements of gBlocks Fragment synthetic mutant template 2x-diluted into a constant background of 100 ng WT DNA demonstrates detection of mutant template present at frequencies of less than 0.05%. The assay sensitivity is approximated by the red dotted line; B, CRISPR- and TALEN-induced point mutations were detected in edited samples at frequencies less than 1%.
A. GRN R493X: NHEJ Quantification by Drop-Off Assay
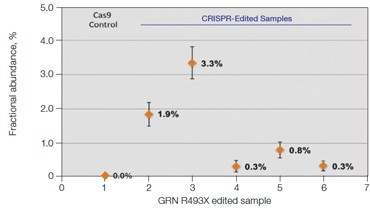
B.GRN R493X: NHEJ Fractional Abundance in Edited Samples
C. GRN R493X: Dilution Series
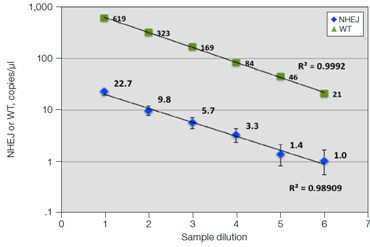
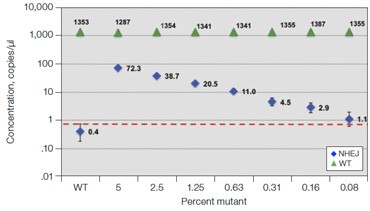
Fig. 6. Quantification of NHEJ alleles in synthetic mutant templates and edited HEK293T cells. A, two merged well analyses of 100 ng of WT gDNA per well with 2x serial dilution of synthetic 1 bp NHEJ deletion gBlocks Fragment template at the GRN locus demonstrates detection down to 0.1%; B, CRISPRinduced point mutations in HEK293T cells can be detected when present at frequencies as low as 0.3%; C, linearity of NHEJ detection across a 2x dilution series in an edited HEK293T sample.
High-Sensitivity Quantification of NHEJ Alleles
Frontotemporal dementia is a neurodegenerative disease caused by mutations in the progranulin gene GRN (OMIM: 607485). To detect NHEJ indels generated by CRISPR/Cas9 we designed a series of drop-off assays. A one base pair deletion in synthetic templates was detected down to a fractional abundance of approximately 0.1% (Figure 6A). In this experiment,
100 ng WT DNA per well was spiked with a 2x serial dilution of a synthetic gBlocks Fragment DNA template containing a one base pair deletion at the predicted Cas9 cut site. Similar sensitivity was observed on a synthetic gBlocks Fragment DNA template with a one base pair insertion (data not shown). The drop-off assay was then used on HEK293T cell samples
treated with editing reagents, showing detection of NHEJ events present at frequencies as low as 0.3% (Figure 6B).
Linearity of measurements of NHEJ events in HEK293T cells over a 2x dilution series is shown for one sample (Figure 6C), demonstrating the ability of this assay to consistently quantify genome editing events across a range of sample inputs. A recent publication confirmed the accuracy of this approach by using drop-off assays in Droplet Digital PCR to quantify NHEJ alleles in TALEN-edited T cells, demonstrating strong concordance to NGS and flow cytometry data (Mock et al. 2015).
Quantification of Cut Site Usage and Bias in Nickase-Edited Samples
Advanced computational tools have been developed that identify candidate gRNAs for efficient targeting of CRISPR-based nucleases. However, there has been limited empirical validation of these computational tools and of predicted gRNA efficiency in general. To examine potential bias in gRNA usage by Cas9 nucleases, we compared NHEJ allele generation at two different gRNA sites used for Cas9 nickase–based editing. In this system, paired Cas9 nickases use two proximal gRNA sites to nick a DNA locus targeted for editing (Ran et al. 2013). A nickase-edited sample was assayed with three types of drop-off assay (Figure 7): one with a probe over each nick site (nickase assay) and two using just a single drop-off probe positioned over one of the two nick sites (CRISPR_A and CRISPR_B assays). Note that HEX amplitude varies as a function of the type and number of NHEJ probes present. The nickase assay has 2 NHEJ probes, CRISPR_A and CRISPR_B, and maximal HEX amplitude is roughly the sum of the individual probe amplitudes (Figure 7A). For this reason, an NHEJ event at one site will lose only partial HEX amplitude with the nickase assay (Figure 7A, blue droplet cluster). As seen in the 2-D plot (Figure 7A) and quantitatively (Figure 7B), in this sample gRNA CRISPR_B produces the majority of NHEJ events, suggesting better nicking efficiency at this site compared to the gRNA CRISPR_A site. Similar trends were observed in a second edited sample in which GRN was targeted. These data demonstrate the ability of Droplet Digital PCR to quantify, empirically validate, and compare gRNA efficacy in cells targeted for editing. This approach therefore offers a quantitative readout useful for empirical validation of gRNA site efficacy predictions made by computational tools.
A.
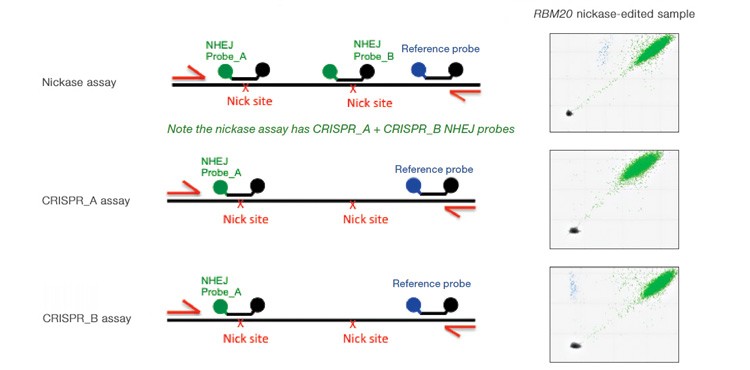
B.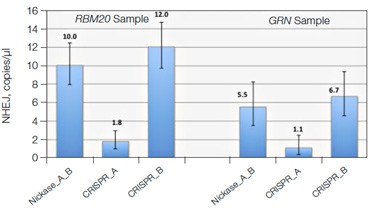
C.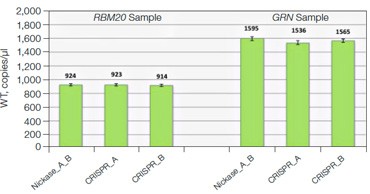
Fig. 7. Quantitative comparison of gRNA usage in nickase-edited samples. A, primer and probe design strategy and representative 2-D plots; B, Using the nickase, CRISPR_A and CRISPR_B assays on RBM20 and GRN edited samples reveals a clear difference in efficacy of different gRNAs, as measured by NHEJ allele generation; C, for each locus, nickase-edited sample were prepared using one sample preparation that was split across three wells for NHEJ quantification. The measurement of WT copies/μl demonstrates, by locus, that equivalent amounts of sample were assayed in each well.
Conclusions
We have shown that Droplet Digital PCR is a powerful technology for ultra-sensitive quantification of NHEJ- and HDR-edited alleles in heterogeneous cell pools. The assay strategies for detection of HDR and NHEJ events described here can detect CRISPR- and TALEN-mediated edits present at frequencies of less than 0.5%. This rapid, cost-effective method is amenable to high throughput and provides a convenient readout for the detection of edited alleles and optimization of editing protocols.
References
Cermak T et al. (2011). Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39, e82.
Cong L et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823.
Miyaoka et al. (2014). Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat Methods 11, 291–293.
Mock et al. (2015). mRNA transfection of a novel TAL effector nuclease (TALEN) facilitates efficient knockout of HIV co-receptor CCR5. Nucleic Acids Res 43, 5660–5571.
Ran FA et al. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389.
Visit bio-rad.com/web/ddPCRgenomeediting for more information.
FAM is a trademark of Applera Corporation. gBlocks is a trademark of Intergrated DNA Technologies Inc. TaqMan is a trademark of Roche Molecular Systems, Inc.
C1000 Thermal Cyclers are covered by one or more of the following U.S. patents or their foreign counterparts owned by Eppendorf AG: U.S. Patent Numbers: 6,767,512 and 7,074,367.
The QX200 Droplet Digital PCR System and/or its use is covered by claims of U.S. patents, and/or pending U.S. and non-U.S. patent applications owned by or under license to Bio-Rad Laboratories, Inc. Purchase of the product includes a limited, non-transferable right under such intellectual property for use of the product for internal research purposes only. No rights are granted for diagnostic uses. No rights are granted for use of the product for commercial applications of any kind, including but not limited to manufacturing, quality control, or commercial services, such as contract services or fee for services. Information concerning a license for such uses can be obtained from Bio-Rad Laboratories. It is the responsibility of the purchaser/end user to acquire any additional intellectual property rights that may be required.