Our first glimpse of CRISPR resulted in a shrug. Setting out to clone and sequence the iap gene in E. coli, Ishino and colleagues at Osaka University stumbled across a strange set of repetitive sequences separated by unrelated short pieces of spacer DNA (Ishino Y et al. 1987) in the 3′ flanking region of iap. This structure was conspicuous enough to merit a mention in their paper, but Ishino could say little more than that no similar structure had been previously described and that the function of this genetic element remained unknown.
Twenty-five years later these mysterious sequences are at the center of some of the biggest hopes and fears that a scientific discovery has ever provoked. From prestigious awards, hundreds of millions of dollars in startup funding, and attention from The New York Times and WIRED Magazine to calls for a moratorium and cautions that CRISPR paves the way for future designer versions of you and me, the reactions are varied. But few of them resemble a shrug.
CRISPR-Cas: A prokaryotic adaptive immune system
Ishino et al were unable to determine a function for the strange repetitive sequences they discovered. But as sequencing technologies advanced similar genetic elements were found in the genomes of distantly related bacteria as well as in many archaea. In 2002 these sequences were given a name: clustered regularly interspaced short palindromic repeats, or CRISPRs. Using in silico analysis of CRISPR loci from various species, Jansen et al (2002) showed that these sequences shared several key features: the previously identified mysterious short spacer sequences flanked by palindromic repeats, a leader sequence that was conserved on the species level, and four CRISPR-associated or cas genes whose homology to exonucleases and helicases suggested a role in DNA metabolism or gene expression.
These findings, combined with reports that CRISPR spacer sequences matched known phage and plasmid sequences (Bolotin et al 2005, Mojica et al 2005, Pourcel 2005), led scientists to hypothesize that CRISPR may be a prokaryotic RNAi-like system that allows bacteria and archaea to defend themselves against invading nucleic acids (Makarova et al 2006, Lillestøl et al 2006).
This hypothesis caught the attention of scientists at Danisco who were battling phages that periodically decimated their yogurt cultures. To test the immune system hypothesis, they infected their Streptococcus thermophilus cultures with known phages. Not surprisingly, most bacteria died. What unlocked today’s CRISPR craze was that the bacteria that did survive had incorporated phage DNA as spacers into their CRISPR loci. When those spacers were removed, the bacteria became phage-sensitive again, supporting the hypothesis that the CRISPR system serves as a form of prokaryotic adaptive immunity (Barrangou 2007). More important for Danisco, this suggested that by inserting sequences of their choosing into CRISPR loci, they could use this bacterial immune system to put an end to productivity- and profit-leaching phage infections.
Turning CRISPRs into tools
Where Danisco saw an opportunity to create phage-resistant strains, other scientists saw the potential for an exquisitely accurate and easily programmable genome editing tool. If CRISPR-Cas could be reprogrammed to cut a cell’s genome at predefined sites, it could be used to disrupt genes or insert DNA sequences through non-homologous end joining (NHEJ) or homology directed repair (HDR).
A key step toward such a genome editing system was taken by the Doudna and Charpentier labs who showed that you can reprogram the S. pyogenes CRISPR-Cas system to cut any desired sequence, in vitro, simply by providing the Cas9 endonuclease and the appropriate guide RNA (gRNA) (Jinek et al 2012). Less than half a year passed before the Zhang lab at MIT used the CRISPR system to edit human and mouse cell lines (Cong et al 2013), showing that this system worked in vivo and across species and domains. Thus began the rapid rise of CRISPR-based genome editing.
How CRISPRs find and destroy their targets
The CRISPR system uses short RNAs, called CRISPR RNAs (crRNAs), to target an endonuclease or endonuclease complex to invading nucleic acids that a prokaryote has previously encountered. Once a foreign piece of DNA has been recognized, it is destroyed.
CRISPR systems identified to date fall into three classes, Types I, II, and III. All CRISPR systems consist of a CRISPR locus, which carries information about past infections, and cas genes, which encode the proteins that mediate CRISPR-based immunity (Figure 1A). CRISPR loci, regardless of type, consist of short (~30 nt) palindromic sequences that flank similarly sized spacer sequences that originate from invading nucleic acids. Though cas genes are usually associated with the CRISPR locus, in some cases they are found elsewhere in the genome.
All CRISPR systems share the same three steps (Figure 1):
Adaption — incorporation of new spacer sequences mediated by Cas1 and Cas2
Expression — transcription of repeat-spacer-repeat sequences into pre-CRISPR RNAs (pre-crRNAs) followed by processing of these sequences into mature crRNAs by type-specific Cas proteins
Interference —crRNA-mediated identification and destruction of invading nucleic acids
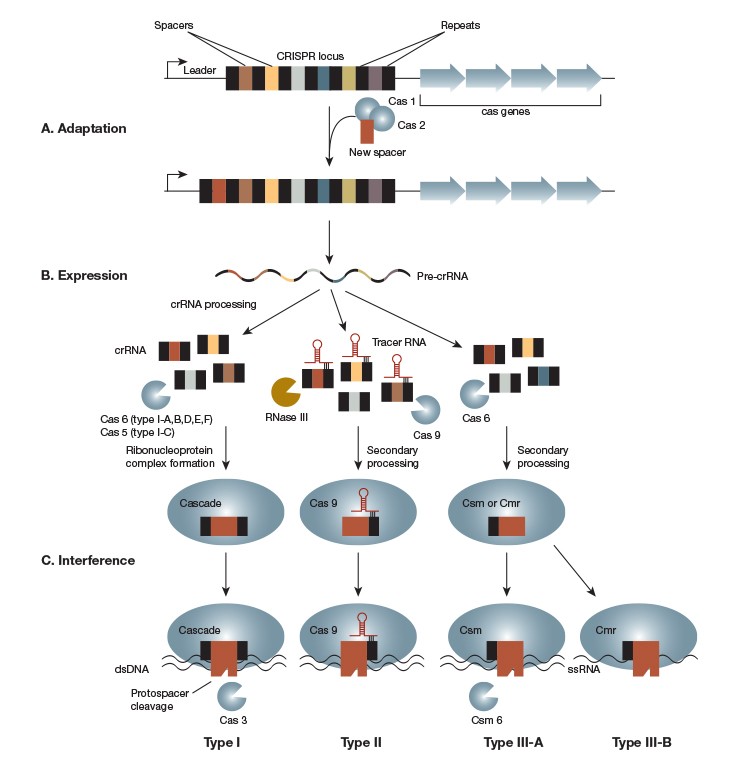
Adapted from Weshra ER et al. (2014)
Fig. 1. The CRISPR-Cas Mechanism. The CRISPR-Cas system consists of a CRISPR locus and several CRISPR-associated or cas genes. New spacer sequences are added to the CRISPR locus by Cas1 and Cas2 as a prokaryote encounters viruses, plasmids, or other invading mobile genetic elements (A). The CRISPR locus is constitutively transcribed into a pre-crRNA that is processed in a CRISPR-type–specific manner. The resulting crRNA associates with type-specific Cas proteins to form an RNA-guided endonuclease complex (B). Foreign DNA is recognized by this complex through its complementarity to the crRNA. Recognition leads to cleavage of the target sequence, referred to as a protospacer, by the endonuclease or endonuclease complex (C). CRISPR systems are categorized into Type I, II, or III based on mechanistic differences in the expression and interference steps. Type I, Type II, and Type III-A systems target double-stranded DNA. Type III-B, however, targets RNA.
The three types of CRISPR systems differ in their processing and interference steps and in their requirements for cleavage (Figure 1A and 1B), a fact that provides us with a treasure trove of CRISPR functionalities. By choosing CRISPR systems from different species, specificity can be altered and CRISPR can even be used to target RNA.
Using CRISPR to edit genomes
To use a system programmed to destroy foreign DNA to introduce specific mutations or DNA sequences into target genomes, CRISPR genome editing technology takes advantage of DNA repair.
crRNAs guide Cas9 proteins to target sequences, which are recognized through their complementarity to the crRNA. The Cas9 nuclease then introduces a double-strand break in the target DNA. This break can be repaired through two pathways — non-homologous end-joining (NHEJ) or homology-directed repair (HDR). The outcome can be either accurate repair or introduction of mutations.
NHEJ may result in small insertions or deletions that can inactivate the target gene. By using two crRNAs, larger deletions can be generated, and if sites on two different chromosomes are targeted, NHEJ can result in chromosomal translocations and inversion.
HDR can be exploited to insert pieces of foreign DNA by providing donor DNA. This can be used to generate conditional alleles, tag proteins, insert specific mutations, or correct existing mutations (Harrison et al 2014). By using a nickase mutant of Cas9 that cuts only one strand of the double-stranded DNA, single-strand breaks are preferentially generated and repair can be biased toward HDR.
Catalytically dead versions of Cas9 (dCas9), on the other hand, can be used as RNA-guided DNA binding proteins that extend CRISPR applications far beyond genome editing. By fusing dCas9 to a transcriptional activator and using a guide RNA (gRNA) that targets the promoter region of a gene, the CRISPR system can be used to activate or enhance transcription (Gilbert et al 2013). Similarly, dCas9 fused to a repressor can be used to downregulate a gene (Gilbert et al 2013). When fused to a fluorescent reporter, dCas9 serves as a beacon that allows visualization of genomic loci of interest (Chen et al 2013).
Making CRISPR work in eukaryotes
Although CRISPR is a prokaryotic system, part of its rapid rise as the genome-editing technology of choice is that it has been adapted to work in almost any model organism, from zebrafish to rice and monkey.
The Type II CRISPR-Cas9 system has been the system of choice for most current genome-editing applications because it is the simplest system; unlike Type I and Type III systems, it requires only a single protein, Cas9, for crRNA maturation, targeting, and target DNA cleavage. This system requires an additional transactivating CRISPR RNA (tracrRNA) for maturation and targeting, but the tracrRNA and crRNA can be combined into a single gRNA that can be custom designed to target almost any sequence (Figure 2) (Jinek et al 2012). Eliminating the need for the RNase III tracrRNA:pre-crRNA processing step means that this system can easily be used in both prokaryotes and eukaryotes. Gene editing in eukaryotes can thus be achieved simply by providing Cas9 and the appropriate gRNA.
A.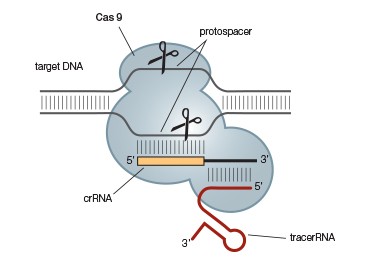
B.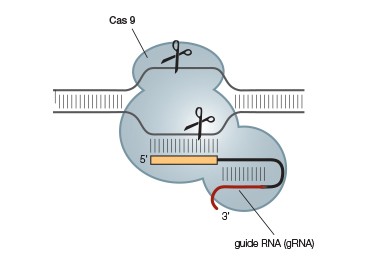
Fig. 2. A simplified CRISPR-Cas9 system. The CRISPR-Cas9 system (A) can be further simplified by combining the required tracrRNA and crRNA into a single guide RNA (gRNA) (B). This allows gene editing of eukaryotes and prokaryotes by simply providing Cas9 and an appropriate gRNA.
CRISPR’s near exponential growth
CRISPR is by no means the first genome editing tool, but it is by far the simplest, fastest, and cheapest, which is why it has already outgrown techniques such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) that have been available for a much longer time.
Both ZFNs and TALENs are protein-based editing tools. Like CRISPRs, they are programmable. But for each new target a new protein has to be engineered, which is a time-consuming, laborious, and, in the case of ZFNs, expensive process.
CRISPR, on the other hand, allows you to target a new gene or generate a new allele with minimal effort: simply design a new gRNA sequence using free algorithms and place your order online. A few days later a plasmid arrives that allows you to edit the genome of your favorite model organism. Because gRNAs are short sequences, the cost of genome editing is minimal — for the price of a few primers you have a gRNA to make the mutation of your choice. CRISPR is thus a simpler and more cost-effective way to generate mutations at rates and in organisms that were previously unimaginable.
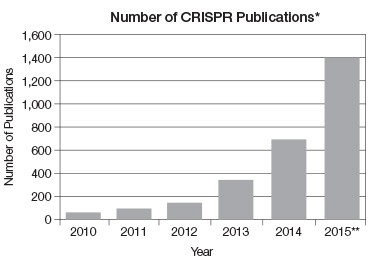
* Based on PubMed data. Includes reviews and primary literature.
** Extrapolated from current number of publications.
Fig 3. The number of CRISPR publications per year has grown almost exponentially.
From pest-resistant wheat to healthier red blood cells
It is thus no surprise that new CRISPR applications are published on a weekly basis. From engineering powdery mildew resistant bread wheat (Wang et al 2014) to using CRISPR multiplexing to generate mice homozygous for mutations in multiple genes within weeks instead of years (Bolcun-Filas et al 2014), it seems that CRISPR is turning genome editing into a simple game of cut and paste and accelerating the pace of research and discovery in an unprecedented way.
CRISPR also open the door to correcting mutations that result in human disease. This is not a new idea — Sangamo Biosciences of Richmond, CA has just completed a phase 2 clinical trial for its ZFN-mediated HIV treatment — but the hope is that the simplicity and affordability of the CRISPR system will accelerate the path to using genome editing to cure human disease. Several companies have been founded on the basis of CRISPR, and together these startups have raised more than $170 million to date (Lauerman and Chen 2015).
And there is good reason to believe that CRISPR will replace other editing technologies. An early study in mice showed that CRISPRs could be used to correct deleterious mutations in vivo: Wu et al (2013) corrected a mutation in the Crygc gene, thereby curing mice of cataracts. Scientists at University of California, San Francisco recently used CRISPR to correct β-thalassemia–causing mutations in human induced pluripotent stem cells (iPSCs) (Xie et al 2014), bringing us one step closer to using edited patient-derived iPSCs for stem cell therapies.
Eliminating off-target edits
One of the hurdles to clinical applications is that CRISPR brings with it some of the same concerns around specificity as other genome-editing techniques. If it is to be used in the clinic, how can we ensure that no off-target edits are made? This has resulted in a call to better understand CRISPR biology before it is used in human therapeutics and has initiated a flurry of research into engineering CRISPR systems with greater specificity.
The current CRISPR system of choice is the CRISPR-Cas9 system from S. pyogenes. This and other Type II systems require a specific 3–9 nt sequence upstream of the target DNA sequence, called the protospacer adjacent motif (PAM), for cleavage. Cas9 systems from other species have PAMs with higher stringency and could thus prove to be part of the puzzle of increasing CRISPR specificity. Another approach has been to better understand the Cas9 enzyme in order to engineer it to be more specific (Kleinstiver et al 2015).
Reinventing how genes are inherited
But specificity is a minor concern when it comes to CRISPR, and one believed to be solvable. It is the power of having discovered the biological version of cut and paste that is creating concerns that, at least at this juncture, nobody has been able to adequately address.
Two studies published earlier this year left the scientific community in both shock and awe. Groups at the University of California, San Diego and Harvard Medical School used CRISPR to turn the theoretical concept of gene drive into a reality. Austin Burt of the Imperial College London proposed the concept of gene drive in 2013 based on naturally occurring selfish genes that are inherited in yeast in an unusual manner. Rather than being passed on following the rules of Mendelian genetics, where each allele has a one in four chance of being transmitted to offspring, selfish genes are passed on to progeny at a much higher rate and could thus take over a population in a very short time.
Using CRISPR, Gantz and Bier of UC San Diego introduced a mutation in Drosophila that transmitted itself to 97% of the progeny (Gantz and Bier 2015). Beyond providing us with a tool to generate new disease models in a fraction of the time of other techniques, gene drive makes it much easier to introduce deleterious mutations that would otherwise be selected against. George Church’s lab at Harvard published similar work earlier this year using CRISPR-based gene drive to bias inheritance in yeast (DiCarlo et al 2015).
The benefits of such a system are clear — creating model systems for disease caused by recessive genes would be greatly simplified and gene drive could be leveraged to eradicate, for example, malaria-causing mosquitos in a single season (Bohannon 2015).
The dangers, however, lie in the unknown. Beyond discussion of whether it is ethically permissible to wipe out an entire population in a matter of months, many believe we do not understand ecosystems well enough to allow such an undertaking. And although Church’s group included genetic precautions meant to prevent the spread of his CRISPR gene drive, and both groups carried out their research in environments built to contain their genetically modified organisms, many in the scientific community point out the need for regulations around what constitutes safe gene drive research and some question whether gene drive experiments should continue.
The National Academy of Sciences has responded to these growing concerns by convening a committee to develop recommendations for responsible conduct of gene drive research. This committee met for the first time on July 30, 2015 and will work to identify techniques for reducing the risks associated with gene drive experiments.
Editing human embryos
This May a study was published that many scientists had dreaded — CRISPR had been used to edit human embryos (Liang et al 2015).
Unlike somatic mutations, mutations made in the germ line will be present in all cells of our body and passed on to our progeny, permanently integrating them into our collective human genome. One can argue that in gene therapy applications we are merely regenerating a wild-type sequence, not generating new genetic material. But part of the concern around CRISPR is that its ease of use and affordability, and differences in human germ line editing regulations in different countries, make it extremely challenging to ensure that correcting deleterious mutations will be CRISPR’s only use.
Another concern, much like that surrounding gene drive, is whether we truly understand biology well enough to intervene in such a drastic way. In a given individual the mutation we are seeking to correct could be a compensatory mutation, a mutation that prevents another, more severe disease. Similarly, being a carrier for one debilitating disease serves in some cases to protect us from another, as with sickle cell anemia and malaria. Correcting such mutations in the germ line could have devastating and irrevocable consequences, some scientists point out.
And then there is the concern about whether we should have a direct hand in our own evolution — should we speed evolution and bend it to what we believe will lead to a better human being? For some, however, the worry is not about speeding evolution. On the contrary, they fear that rather than accelerating or directing evolution, we could be reducing diversity and thus creating genetic bottlenecks.
Pausing to define a prudent path for CRISPR research
These and many similar concerns have led scientists who are using CRISPR and other genome-editing techniques to call not only for dialogue but also for action. From a flood of commentaries and letters in journals such as Science and Nature to a suggested voluntary moratorium on human germ line editing proposed by Sangamo’s CEO, the scientific community is calling for action.
This dialogue has grown beyond the scientific community, as the potential impacts of CRISPR are being outlined by media outlets such as The New York Times, Business Insider, and WIRED Magazine.
This past January experts in genome editing, bioethics, and law, as well as many of the researchers who pioneered CRISPR technology, attended the Innovative Genomics Initiative Forum on Bioethics in Napa, CA to discuss the future of CRISPR research. Their recommendations, published in the journal Science, (1) strongly discourage human germ line modifications until we better understand the implications of such experiments, (2) call for the creation of a forum to discuss and advise on these implications, (3) encourage transparency in CRISPR research to determine the efficacy and specificity of CRISPR-based genome editing, and (4) ask for the formation of a global body that brings together scientists, bioethicists, lawyers, representatives of the public, and relevant government agencies to recommend policies when appropriate (Baltimore et al 2015).
The National Institutes of Health’s response is to not fund any research that involves editing of human embryos.
The future of CRISPR
We are clearly at only the beginning of the conversation about the future of CRISPR research, the conversation about its incredible potential and about whether and how regulations limiting CRISPR use could be enforced. We cannot unknow what is known, we cannot unpublish papers, or collectively agree to unremember CRISPR technology. And although most agree that a healthy dose of fear is appropriate, CRISPR’s potential to answer questions about biology at a pace none of us could have imagined and its ability to improve our lives lead many to point out that CRISPR should elicit more hope and excitement than dread and that we should not close the door on such a powerful technology.
Bioradiations would like to thank Yan Wang and Rongdian Fu for technical comments and resources.
References
Baltimore D et al. (2015). A prudent path forward for genomic engineering and germline gene modification. Science 348, 36–38.
Barrangou R et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1,709–1,712.
Bohannon J (2015). Biologists devise invasion plan for mutations. Science 347, 1,300.
Bolcun-Filas E et al. (2014). Reversal of female infertility by Chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science 343, 533–536.
Bolotin A et al. (2005). Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151, 2,551–2,561.
Chen B et al. (2013). Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1,479–1,491.
Cong L et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823.
Gilbert LA et al. (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451.
Harrison MM et al. (2014). A CRISPR view of development. Genes Dev 28, 1,859–1,872.
Ishino Y et al. (1987). Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169, 5,429–5,433.
Jansen R et al. (2002). Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43, 1,565–1,575.
Jinek M et al. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821.
Kleinstiver BP et al. (2015). Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–5.
Lauerman J and Chen C. (2015) The promise and peril of CRISPR. Bloomberg Businessweek. http://www.bloomberg.com/news/articles/2015-06-25/the-promise-and-peril-of-crispr, accessed 7/30/15.
Lillestøl RK et al. (2006). A putative viral defence mechanism in archaeal cells. Archaea 2, 59–72.
Liang P et al. (2015). CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 6, 363372.
Makarova KS et al. (2006). A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1, 7.
Mojica FJ et al. (2005). Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60, 174–182.
Pourcel C et al. (2005). CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151, 653–663.
Sampson TR and Weiss DS (2014). Exploiting CRISPR/Cas systems for biotechnology. Bioessays 36, 34–38.
Wang Y et al. (2014). Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol 32, 947–951.
Weshra ER et al. (2014). CRISPR-Cas systems: beyond adaptive immunity. Nature Reviews Microbiology 12, 317-26.
Wu Y et al. (2013). Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 13, 659–662.
Xie F et al. (2014). Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res 24, 1,526–1,533.