Abstract
The near complete removal of host cell protein (HCP) impurities from biologics (therapeutic protein drugs) is essential for patient safety and treatment efficacy. Hence, biologics are routinely monitored for levels of residual HCPs by a highly sensitive ELISA/immunoassay with a polyclonal antibody reagent raised against a wide range of potential HCP impurities (anti-HCP antibodies). To ensure accuracy of the ELISA, the range of HCP impurities detectable by the anti-HCP antibodies must be evaluated. Although 2-D electrophoresis (2-DE) and western blotting (described in detail in Bio-Rad’s Applications & Technologies section) is the gold standard method for evaluating anti-HCP antibodies, its adoption is hampered by its perceived lack of reproducibility and labor/time-intensive nature of the standard workflow. Here, we demonstrate an improved, optimized workflow for streamlined and highly reliable, 2-DE and western blotting-based evaluation of anti-HCP antibodies.
Introduction
Most therapeutic proteins are produced by cultured cells that have been engineered through recombinant DNA technology to produce the relevant protein. The protein is either produced in the host cell or secreted into the medium. In the case of intracellular protein production, the host cells are harvested and lysed, and the protein of interest is purified from the lysate. In the case of secreted protein, the protein of interest is purified from the culture medium in which the cells have been grown. In either case, the starting material for purification of the protein of interest contains other proteins from the host cell line, and these so-called host cell proteins (HCPs) are potential impurities in the final drug substance. HCP impurities can induce an immune response in the patient, so their presence needs to be minimized. Validation and quality control testing of any therapeutic protein needs to include a test to show that HCP contamination is at a low, permissible level.
HCP contaminants can have an undesirable effect even at very low concentration and in general, the only sufficiently sensitive assay methodology for HCP is immunochemical. The presence of HCP is usually assayed by ELISA using a polyclonal antibody mixture that has been developed to have reactivity against a wide range of potential HCPs. Development of the polyclonal antibody requires optimization of the number of potential targets recognized, and the specificity of the assay needs to be fully validated prior to adoption.
A recommended method for this validation involves two dimensional (2-D) electrophoretic separation followed by western blotting (Savino et al. 2011, Wang et al. 2009). This technique displays the proteins in a representative HCP sample as an array of hundreds to thousands of individual spots. Western blotting determines which of those proteins are recognized by the polyclonal antibody. This allows the calculation of a match rate or coverage score corresponding to the ratio of the number of proteins recognized by the antibody to the total number of proteins detectable in the sample. Although this method allows a straightforward determination of the range of reactivity of the antibody mixture, adoption of this method has been hindered by the perceived lack of sensitivity, lack of reproducibility, and overall laborious nature of the 2-D electrophoresis and blotting workflow.
Tools that simplify, standardize, and accelerate 2-D electrophoresis and blotting have become available. Advanced electrophoresis and blotting instrumentation, stable and reproducible precast gels, and next-generation imaging and image analysis software all allow this assay to be performed in a reproducible manner in a relatively short time. We have undertaken to demonstrate this by generating coverage scores for a commercially available anti-HCP antibody against samples derived from CHO cells, a common cell line used in the production of protein therapeutics.
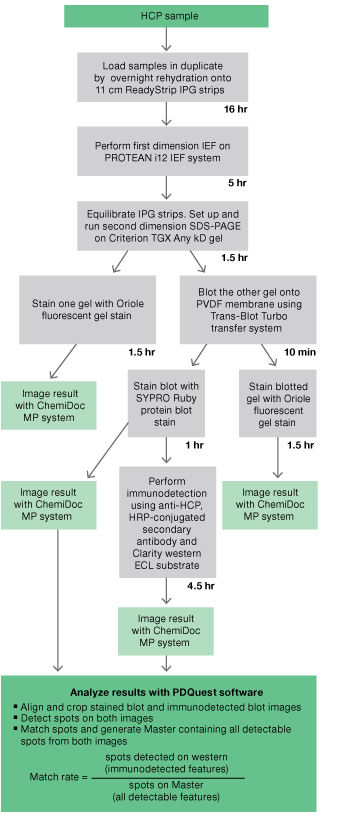
Fig 1. Workflow for evaluation of antibodies developed fordetection of host cell proteins.
Materials and Methods
Materials
ReadyPrep™ 2-D cleanup kit, ReadyPrep 2-D rehydration/sample buffer, ReadyStrip™ IPG strips, Criterion™ gels, Trans-Blot® Turbo™ PVDF transfer packs, Oriole™ fluorescent gel stain, SYPRO Ruby protein blot stain, blotting grade blocker, Clarity™ western ECL substrate, and associated buffers and standards were from Bio-Rad Laboratories. Ultrafiltration cartridges were from Sartorius (Bohemia, NY). The anti-HCP antiserum was a goat IgG fraction raised against CHO cell HCPs recovered from conditioned growth media (Cygnus Technologies, Southport, NC). HRPconjugated bovine anti-goat antibody was from Santa Cruz Biotechnology (Dallas, TX).
Samples
CHO-K1 cells were grown as adherent cultures and adapted to a medium consisting of 95% protein-free defined medium (Gibco, Life Technologies, Grand Island, NY) and 5% Ham’s medium containing 10% fetal bovine serum (0.5% fetal bovine serum total). Two days prior to harvest, the cells were washed with PBS and transferred to 100% protein-free defined medium. The CHO cell secreted protein sample was prepared by ~100-fold centrifugal concentration (10,000 MWCO), treated with the 2-D cleanup kit, and resuspended
2-D Electrophoresis
First dimensions were carried out using the PROTEAN® i12™ IEF cell. Samples (150 μg protein) were applied to 11 cm pH 3–10 NL strips by overnight rehydration. The CHO cell secreted protein sample was applied in a solution comprising 8 M urea, 4% CHAPS, 40 mM DTT, and 0.2% Bio-Lyte® 3/10 carrier ampholyte. The CHO cell lysate was applied in the same solution with 7 M urea, 2 M thiourea in place of 8 M urea. First-dimension IEF was run in the “gel-side up” mode using the pre-programmed “gradual” protocol. The second dimensions were run on Criterion™ TGX Any kD™ gels using the Criterion™ Dodeca™ cell.
More experimental details may be found in the 2-D Electrophoresis Workflow How-To Guide, bulletin 2651.
Blotting and Staining
Gels were either stained with Oriole fluorescent gel stain or transferred onto PVDF membrane using the Trans-Blot Turbo system. Following transfer, the PVDF membranes were stained with SYPRO Ruby protein blot stain to visualize transferred protein. The blotted gel was stained with Oriole stain post-transfer as well to visualize any protein remaining in the gel.
Immunodetection
Immunodetection was carried out directly following imaging of the SYPRO Ruby-stained blot. The membranes were blocked using 5% blotting grade blocker (nonfat dry milk) in Tris-buffered saline containing 0.05% Tween 20 (TTBS) for 1 hr with agitation. Blots were subsequently incubated in primary anti-HCP antibody diluted 1:150 in the same solution used for blocking, then washed in TTBS five times for 5 min each. The blot was incubated in HRP-conjugated bovine anti-goat secondary antibody diluted 1:3000 in TTBS for 1 hr, then washed six times for 5 min each. Chemiluminescent development was performed with the Clarity western ECL substrate.
More experimental details may be found in the Protein Blotting Guide, bulletin 2895.
Imaging and Analysis
Fluorescence imaging of stained gels and blots, as well as chemiluminescent imaging of immunodetected blots, was performed with the ChemiDoc™ MP imager. Image analysis was performed using PDQuest™ 2-D analysis software. False color image overlays were prepared using Adobe Photoshop.
Workflow
The workflow for evaluation of anti-HCP antisera is outlined in Figure 1. The HCP sample was loaded in duplicate and processed through all the steps of 2-D electrophoresis.
One gel was stained with a rapid, sensitive fluorescent stain (Oriole) and the other was transblotted onto PVDF. Following transfer, the blotted gel was also stained with Oriole stain.
All of the steps up until this point (overnight sample loading followed by first-dimension IEF, SDS-PAGE second dimension, fluorescent staining, and transfer to PVDF membrane) were conducted over the course of 24 hours. The blotted membrane was rinsed with water, allowed to dry, and stored overnight.
Staining of the blots for total protein (with SYPRO Ruby stain), immunodetection, chemiluminescent development, and imaging were all conducted over the course of the second day.
The staining of a duplicate gel with Oriole stain, and the staining of the blotted gel with Oriole stain following transfer, allowed a visual verification of transfer efficiency. Staining the blot for total protein with SYPRO Ruby stain allowed further verification of transfer efficiency, and simplified analysis by allowing the comparison of total protein and immunodetected protein patterns generated from the same blot.
PDQuest software was used to generate match rates between the images of the stained blot and the immunoblot. The detailed procedure is covered in the appendix.
Results
2-D Spot Pattern Reproducibility
CHO cell secreted protein samples were analyzed in triplicate by 2-D electrophoresis and Oriole staining, as described, in order to evaluate the reproducibility of the spot patterns generated and the number of identifiable protein species. The three results were qualitatively very similar to one another and allowed the identification of 526–580 individual protein spots with 65–72% of them able to be matched between gels by software analysis (Figure 2).

Fig 2. 2-D spot pattern reproducibility. CHO cell secreted protein was separated by 2-D electrophoresis. The gels were stained with Oriole stain and counting and matching were performed using PDQuest software. There were 380 spots that could be matched among all three gels. The overall match rate was 65–72%.
2-D Western Blots of CHO Cell Secreted Protein Probed with Anti-CHO HCP
The workflow described was applied to a sample derived from CHO cell secreted protein (Figure 3). The 2-D pattern on the stained blot is very similar to the replicate stained gel image, indicating that the transfer introduced little mass or pI bias. Very little protein was evident in the post-transfer stained gel, indicating good transfer efficiency. Western blotting detected a number of proteins recognized by the antibody and a false color overlay of the SYPRO Ruby stain-visualized total protein image and the western blot image shows the correspondence between the two images (Figure 4).
/2013/05/2-d-western-blotting-steps-of-cho-cell-lysate.jpg” alt=”2-D western blotting steps for CHO cell secreted proteins” width=”696″ height=”127″ class=”aligncenter size-full wp-image-1418″ />
Fig 3. CHO cell secreted protein probed with anti-CHO HCP — stained gels, stained blot and western blot. CHO cell secreted protein was separated in duplicate by 2-D electrophoresis. A, one gel was stained with Oriole fluorescent gel stain; B, the second gel was transferred to PVDF membrane and stained with Oriole grain following transfer; C, the PVDF membrane was stained with SYPRO Ruby blot stain to visualize total protein; D, following staining, the blot was immunodetected with anti-CHO HCP and visualized by chemiluminescence.

Fig 4. CHO cell secreted protein probed with anti-CHO HCP and false color overlay of stained blot with western blot image.The stained blot and western blot images from Figure 3 were overlaid. SYPRO Ruby stain-visualized total protein is shown in Blue. Protein immunodetected with anti-CHO HCP is shown in Red.
2-D Western Blots of CHO Cell Lysate Probed with Anti-CHO HCP
The same workflow was applied to a CHO cell lysate (Figures 5 and 6).
Software Matching between 2-D Images and Generation of Match Rates
PDQuest software was applied to the stained blot and western blot images to detect spots and match them between images. A match rate was calculated that corresponded to the ratio of spots detected on the western blot to the total number of spots detectable by both methods (Figures 6 and 7, Table 1).
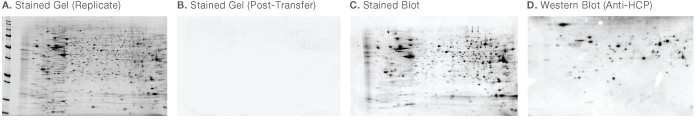
Fig 5. CHO cell lysate probed with anti-CHO HCP — stained gels, stained blot, and western blot. A, one gel was stained with Oriole fluorescent gel stain; B, the second gel was transferred to PVDF membrane and stained with Oriole following transfer; C, the PVDF membrane was stained with SYPRO Ruby blot stain to visualize total protein; D, following staining, the blot was immunodetected with anti-CHO HCP and visualized by chemiluminescence.
Table 1. Generation of match rate from PDQuest data. PDQuest software was used to analyze images of stained blots and western blots of CHO cell-derived samples. Details of the analysis are in the appendix.
| Secreted Protein | Lysate | |
| Number of features detected on stained blot (total protein) | 354 | 826 |
| Number of features detected with anti-HCP (western blot) | 204 | 173 |
| Number of features matched between the two methods | 128 | 154 |
| Total number of features detected between the two methods | 430 | 845 |
| Match rate (ratio of features detected with anti-HCP to total detected features) | 47% | 20% |
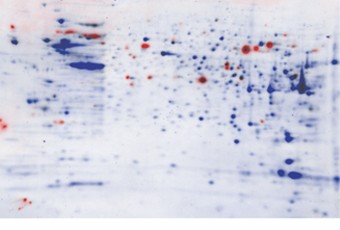
Fig 6. CHO cell lysate probed with anti-CHO HCP and false color overlay of stained blot with western blot image.The stained blot and western blot images from Figure 3 were overlaid. The stained blot and western blot images from Figure 4 were overlaid. SYPRO Ruby stain-visualized total protein is shown in Blue. Protein immunodetedted with anti-CHO HCP is shown in Red.
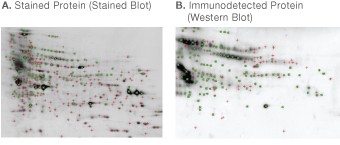
Fig 7. Software matching between stained blot (A) and western blot (B) of CHO cell secreted protein. PDQuest software was used to detect and match spots. Detected spots are indicated by the symbol (+), spots matched between the two images are indicated by the symbol (×).
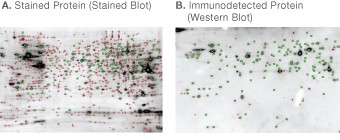
Fig 8. Software matching between stained blot (A) and western blot (B) of CHO cell lysate. PDQuest software was used to detect and match spots. Detected spots are indicated by the symbol (+). Spots matched between the two images are indicated by the symbol (×).
Discussion
The workflow described here utilizes various innovations that have brought speed, simplicity, and reproducibility to 2-D electrophoresis and western blotting. All of the steps have been optimized to give high quality results in the shortest possible time. The net result was a procedure than can be comfortably performed over the course of two days.
The 2-D cleanup kit selectively and quantitatively precipitates proteins from complex samples and allows them to be resuspended in a solution compatible with first-dimension IEF. In this case, use of this kit was critical in producing a high-quality 2-D separation from proteins derived from concentrated cell culture supernatant.
Precast immobilized pH gradient (IPG) strips and the PROTEAN i12 IEF system bring reproducibility and convenience to the first dimension.
Criterion TGX AnykD gels allow rapid second-dimension separations while providing the consistency and quality enabled by precast gels. The Criterion gel format is an ideal format for maximizing 2-D gel spot counting while maintaining the relative ease of handling provided by a midi-format gel.
The Trans-Blot Turbo system has been optimized to give effective transfer over a wide mass and pI range (Sun et al. 2012). Transfer can be completed in as little as 7 min.
Oriole stain is an easy-to-use and rapid fluorescent protein stain that gives nanogram sensitivity similar to silver staining. Staining a replicate gel with Oriole stain allowed confirmation of the quality and resolution of the 2-D separation. Staining the gel with Oriole stain following transfer allowed the confirmation that transfer was nearly complete.
The use of SYPRO Ruby blot stain to visualize total protein prior to immunodetection was critical to the success of the workflow. This rapid fluorescent membrane stain allows high sensitivity visualization of transferred protein and does not interfere with downstream immunodetection (Berggren et al 1999). Comparing the 2-D pattern on the blot to the 2-D pattern on the Oriole-stained gel confirmed the lack of any strong bias with respect to mass or pI as a result of transfer. It also allowed very effective software matching between the stained total protein image, and the immunodetected western blot image. Since total protein and immunodetected protein are imaged on the same membrane, the images are in register with each other and software matching does not require image warping or compensation for differences in spot position between images. This results in high confidence matching with relatively little manual intervention. When replicate gels were matched to each other, match rates of 65–72% were obtained despite qualitatively very similar 2-D patterns. This result highlights the significant error potential for calculating match rates between replicate runs that is avoided by performing the analysis on the same membrane.
Chemiluminescent immunodetection is the most sensitive method for visualizing western blots and the Clarity western ECL substrate offers high sensitivity, low background, and exceptionally long signal duration, which results in high consistency between experiments.
The ChemiDoc MP imaging system allows both fluorescent and chemiluminescent images to be captured at the same resolution with the same camera. This further simplifies matching between the fluorescently stained total protein image and the chemiluminescent western blot image.
The western immunodetection protocol used here is a standard method that gives good quality western blot images with relatively little nonspecific cross-reactivity. Higher or lower stringency conditions may be employed by varying the duration and number of wash steps, or by using different blocking reagents. This will affect the number of immunodetected spots, and consequently, the calculated coverage rate. It is important, therefore, to maintain standard conditions when comparing different antibody preparations or different samples.
Optimization of the primary and secondary antibody dilutions should be performed prior to carrying out the workflow. This is most conveniently done by performing the western blot experiment on 1-D gel-separated material. The sample to be probed can be run in several parallel lanes of the same SDS-PAGE gel. Following transfer, the blot is cut into strips corresponding to individual lanes, and immunodetection is performed on each strip using varying concentrations of primary and secondary antibody. In general, one chooses a combination that gives high detection sensitivity of as many bands as possible while minimizing background reactivity and maintaining a good image appearance.
In both samples tested, there were prominent, abundant proteins detected by protein staining that were not apparent in the corresponding western blot. These represent proteins, which, if present as HCP contaminants in a drug substance, might not be detected by an ELISA performed using this antibody. There were also a number of features on each western blot that were not matched to proteins detected by the total protein stain. These probably represent strongly immunogenic proteins whose abundance falls below the threshold of detectability by SYPRO Ruby blot stain.
The anti-CHO antibody tested gave a considerably higher match rate on the secreted protein preparation (47%) than on the lysate (20%). This is consistent with the fact that this antibody was generated using proteins recovered from CHO cell growth medium. The immunodetected proteins present in the lysate may be proteins that were present in the preparation used to generate the antibody due to lysis during cell culture. They may also represent secreted proteins that are also present intracellularly.
The match rate generated by this method depends on a number of variables beyond the sample and the antibody. As noted above, the conditions used for immunodetection can influence the result, as can the method used to stain and visualize total protein. Comparing Figure 2 and Table 1, it is apparent that the same sample gives more identifiable spots on a gel stained with Oriole stain (526–588) than on a blot stained with SYPRO Ruby stain (354). This results from the intrinsically higher sensitivity of the gel stain compared to the blot stain. It is apparent that a more sensitive total protein staining method would result in a lower apparent match rate. The parameters used for spot detection and matching will also influence the match rate. It is important, therefore, to keep these parameters consistent when comparing among antisera or samples, or when optimizing an antiserum to a particular sample type.
References
Berggren K et al. (1999). A luminescent ruthenium complex for ultrasensitive detection of proteins immobilized on membrane supports. Anal Biochem 276, 129-143.
Savino E et al. (2011). Development of an in-house, process-specific ELISA for detecting HCP in a therapeutic antibody, Part 1. BioProcess Intl 9, 38-47.
Sun C et al. (2012). Robust transfer of proteins to membranes across a wide range of pI and mass using the Trans-Blot Turbo system. Bio-Rad Bulletin 6147.
Wang X et al. (2009). Host cell proteins in biologics development: Identification, quantitation and risk assessment. Biotechnol Bioeng 103, 446-458.
Bio-Rad Laboratories, Inc. is licensed by Invitrogen Corporation to sell SYPRO products for research use only, under U.S. patent 5,616,502.
Gibco is a trademark of Life Technologies Corporation.
Photoshop is a trademark of Adobe Systems Incorporated.
SYPRO is a trademark of Invitrogen Coporation.
Tween is a trademark of ICI Americas Inc.>


